本帖最后由 Jack小新 于 2026-4-7 22:13 编辑
导读:一把剪刀剪掉多余的氨基酸,活性位点却丝毫不动 —— 这不是科幻,而是来自 Imperial College London 团队刚刚上传至 bioRxiv 的最新工作。他们用蛋白质语言模型 (PLM)+ 蒙特卡洛采样,把 PETase、Taq DNA 聚合酶等四种明星工业酶压缩到原来的一半甚至更小,同时在计算层面保住了催化活性口袋的几何构型。本文带你逐步拆解这套「酶减重流水线」。
一、为什么要给酶「减肥」?
自然界经过亿万年进化选出来的酶,往往是为了在细胞内环境中同时应对多种任务而设计的 —— 不一定是最小的,也不一定是最适合工业生产的。
对于实际应用而言,酶太大会带来一连串麻烦:
基因递送受限:腺相关病毒 (AAV) 载体对包装基因组大小有严苛限制,太大直接塞不进去;
生产成本高企:更长的序列意味着更慢的发酵、更低的可溶表达量;
工程改造困难:越大的蛋白质,设计空间越难以穷举。
因此,有一个经典问题在蛋白质设计领域一直悬而未决:
给定一个酶和它已知的催化位点几何结构,能折叠成保留该活性位点构型的、最短的氨基酸序列是什么?
这就是 "基序支架化 (motif scaffolding)" 问题在酶工程中的体现。而这篇文章,正是给出了一条基于 PLM 的系统性解答。
二、核心思路:把缩短序列变成一道 "能量最小化" 问题
整个方法的精髓,是把「酶的压缩」重新表述为在一个精心设计的能量景观上进行采样。
能量函数
论文定义了一个包含两项的目标能量函数 (大正则势能):
E=Eembedding+Echemical
Eembedding (嵌入相似能):衡量当前序列中活性位点残基的 PLM 嵌入向量,与野生型 (WT) 对应残基嵌入向量的余弦相似度。这一项确保优化过程中,活性位点的 "语义上下文" 不跑偏;
Echemical (化学势能):惩罚序列总长度超出目标长度的程度,相当于一个让序列不断收缩的 "弹簧力"。
两项合在一起,优化过程的平衡点自然落在:序列足够短,但活性位点的嵌入特征仍与 WT 高度相似 —— 这正好对应于「保住功能、最大化压缩」的目标。
类比统计力学,wchemical⋅μ 扮演的角色等同于大正则系综中的化学势,通过调节它的大小,可以精准控制压缩力度。
优化引擎:BAGEL + 蒙特卡洛
优化在开源软件包 BAGEL 中实现,采用标准蒙特卡洛 (MC) 采样,每一步随机提出替换、插入或删除三类操作 (概率比 70%:15%:15%),用 Metropolis 接受准则决定是否采纳。嵌入计算使用 ESM-2 (650M) 模型。
三、流水线全貌:从序列海洋到实验候选
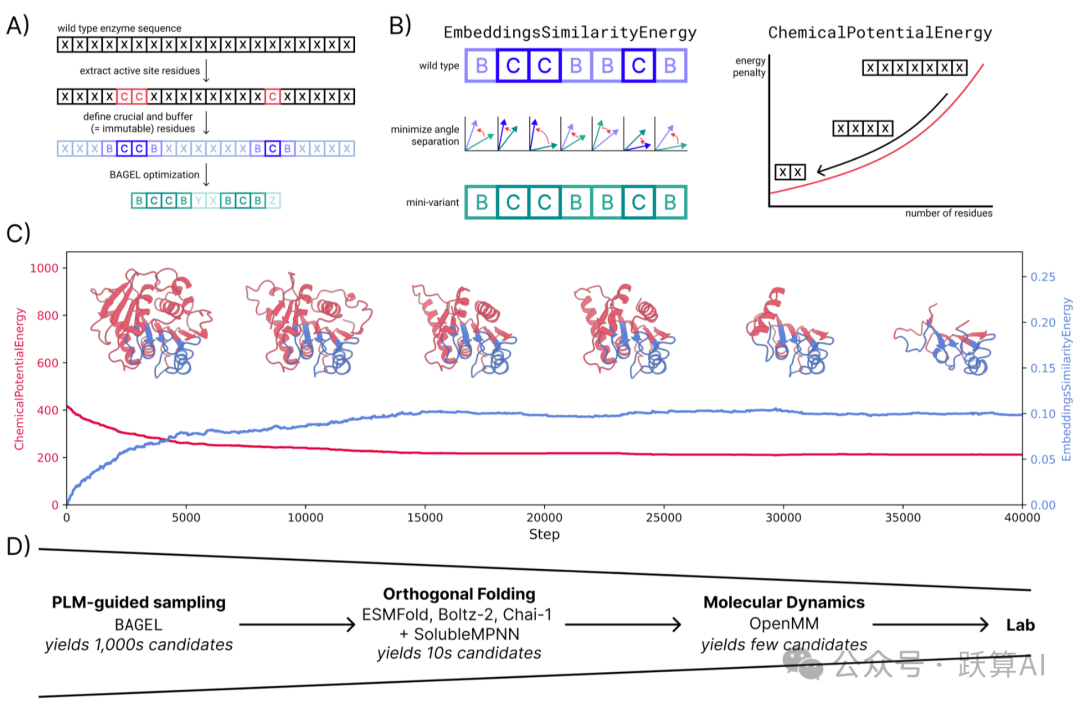
图 1 PLM 引导的酶微型化流水线全览来源:论文 Figure 1
A:流程示意。从 WT 序列中提取关键催化残基(C),以及左右各 4/3 个氨基酸的缓冲 区(B),合称不可变区域(I=B∪C),在优化中固定不动;其余残基作为可变区域, 由 BAGEL 自由探索
B:两个能量项的示意。嵌入相似能通过最小化活性位点残基嵌入向量之间的角度来保护活性口袋语义;化学势能通过对序列长度施加惩罚来驱动序列缩短。
C:一次典型 MC 轨迹的能量演化。Echemical 单调下降直至平台,说明压缩已到极限;Eembedding 从零缓慢爬升后也趋于平稳 —— 两项能量达到新的平衡。
D:漏斗式筛选流程。MC 采样产生数千个候选序列 → 三个折叠模型 (ESMFold / Chai1 / Boltz-2) 筛选出数十个 → 分子动力学 (MD) 模拟进一步过滤,留下少量实验验证候选序列。
四、结果①:化学势越大,压缩越猛,但代价也越大
研究者首先做了权重扫描实验,在五个档位上各运行 5 条独立轨迹,通过 ESMFold 折叠后分析不可变区域的 pLDDT (预测置信度) 和 RMSD (与 WT 的结构偏差)。
规律很直观:化学势越大 → 序列越短 → pLDDT 越低、RMSD 越高。但在较低权重 (≤0.001) 时,PETase、蛋白酶和 Taq 聚合酶这三个酶仍能维持高 pLDDT 和低 RMSD,只有 VioA 出现较明显的连续下滑。
综合权衡,研究者将所有酶统一设定 wchemical=0.001 进行后续生产运行。
图2 化学势权重对酶微型化的影响来源:论文 Figure 2
A:VioA 在不同 wchemical 下的代表性折叠结构。蓝色为不可变区域,红色为可变区域,黑色轮廓为 WT。权重越大,蓝色区域结构越扭曲,说明活性位点已被破坏。
B (上):四种酶的 pLDDT 分布随权重变化情况。较低权重下 petase/protease/taq 分布集中在高置信度区间;vioA 随权重升高整体左移。
B (下):不可变区域 RMSD 分布 (对数坐标)。低权重时 RMSD 主峰紧贴 以下;高权重时整体向 Å 移动,意味着活性口袋几何结构严重偏离 WT。
五、结果②:BAGEL 远胜 "暴力端切"
一个自然的疑问是:直接从 N 端和 C 端往里剪,效果如何?研究者设计了一个确定性基线算法 —— 手动修剪 (manual trimming):
Stage 1:交替从 N 端和 C 端删除残基,直到触及不可变区域;
Stage 2:对内部残基段做轮换删除,直到只剩不可变区域。
对比结果表明,两种方法在端部修剪阶段表现相近;但一旦需要删除内部残基 (Stage 2),BAGEL 的优势立刻凸显 —— 在相同序列长度下,嵌入角度更小、RMSD 更低、pLDDT 更高。这说明 BAGEL 能够通过在可变区域引入替换突变,来「补偿」内部缺失带来的结构扰动,而暴力删除则无能为力。
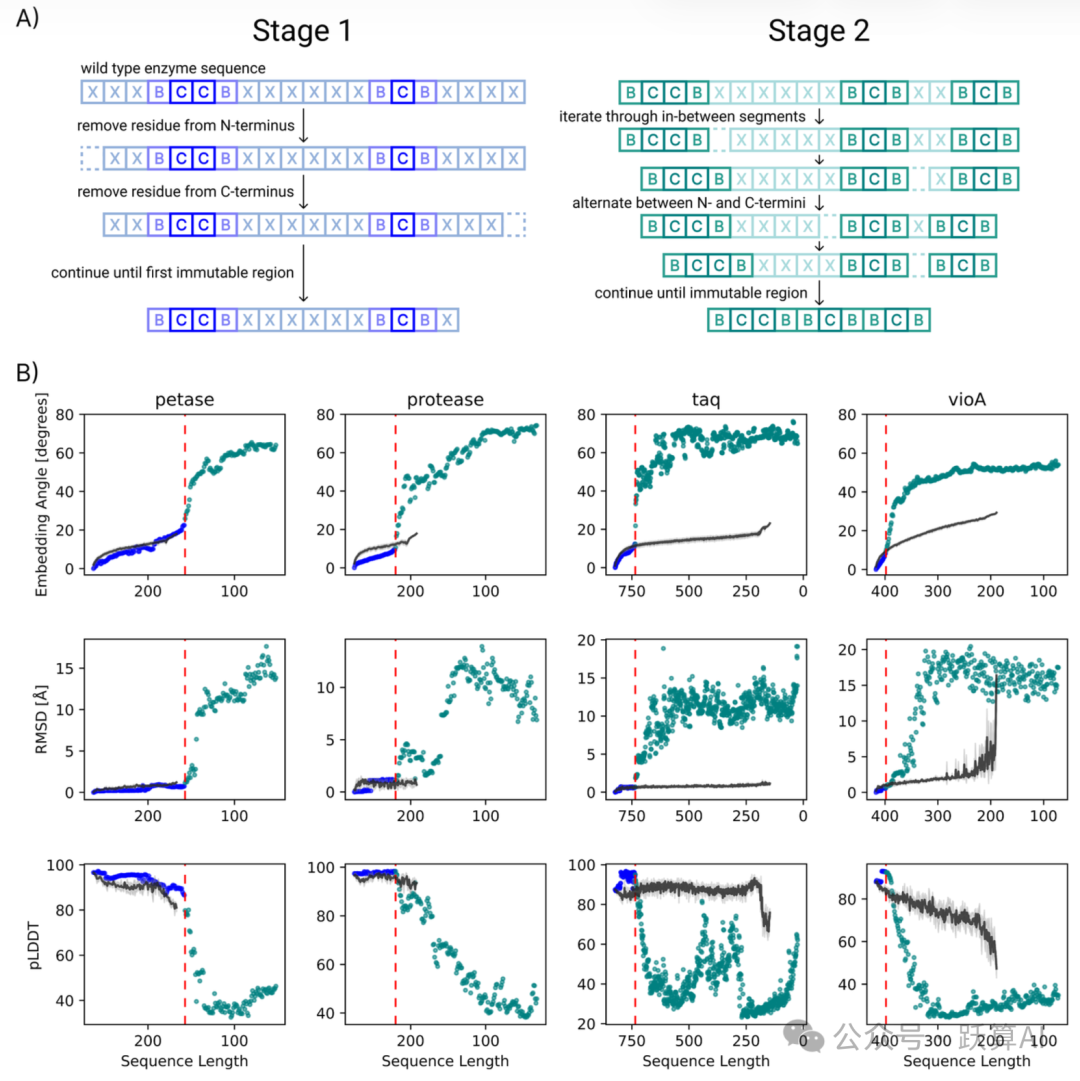
图 3 手动修剪 vs. PLM 引导 BAGEL 优化的对比来源:论文 Figure 3
A:手动修剪两阶段示意图。Stage 1 交替从两端删除 (蓝色),Stage 2 对内部各段循环删除 (青绿色)。
B:四种酶在序列长度压缩过程中的三项指标轨迹 (从左到右序列逐渐缩短)。
第一行:嵌入角度 (度)。BAGEL (黑色点) 在短序列区间的嵌入角度远低于手动修剪轨迹,说明活性位点语义更接近 WT。
第二行:RMSD (Å)。taq 和 vioA 在进入 Stage 2 后手动修剪的 RMSD 急剧飙升,而 BAGEL 保持相对平稳。
第三行:pLDDT。BAGEL 序列在较短长度下仍维持 >80 的高置信度,手动修剪则大幅下跌。
六、结果③:三模型结构共识筛选
为避免 "幻觉折叠"—— 即模型虚报高置信度但结构实际错误的情况 —— 研究者用三个架构迥异的折叠模型对所有候选序列独立折叠:ESMFold、Chai-1、Boltz-2。只有在三个模型中不可变区域 RMSD 均低于阈值的序列,才进入 MD 验证阶段。
主要发现:
· PETase 和 protease 表现最佳,三模型 RMSD 分布高度一致,均集中在 以下,普遍
· Taq 表现中等,ESMFold 和 Chai-1 一致性较好,但 Boltz-2 给出较高 RMSD;
· VioA 偏差最大,三模型之间分歧也最明显,需要放宽 RMSD 阈值至 才能保留足够候选。
最终每种酶筛选出 top-16 候选序列送 MD 验证。
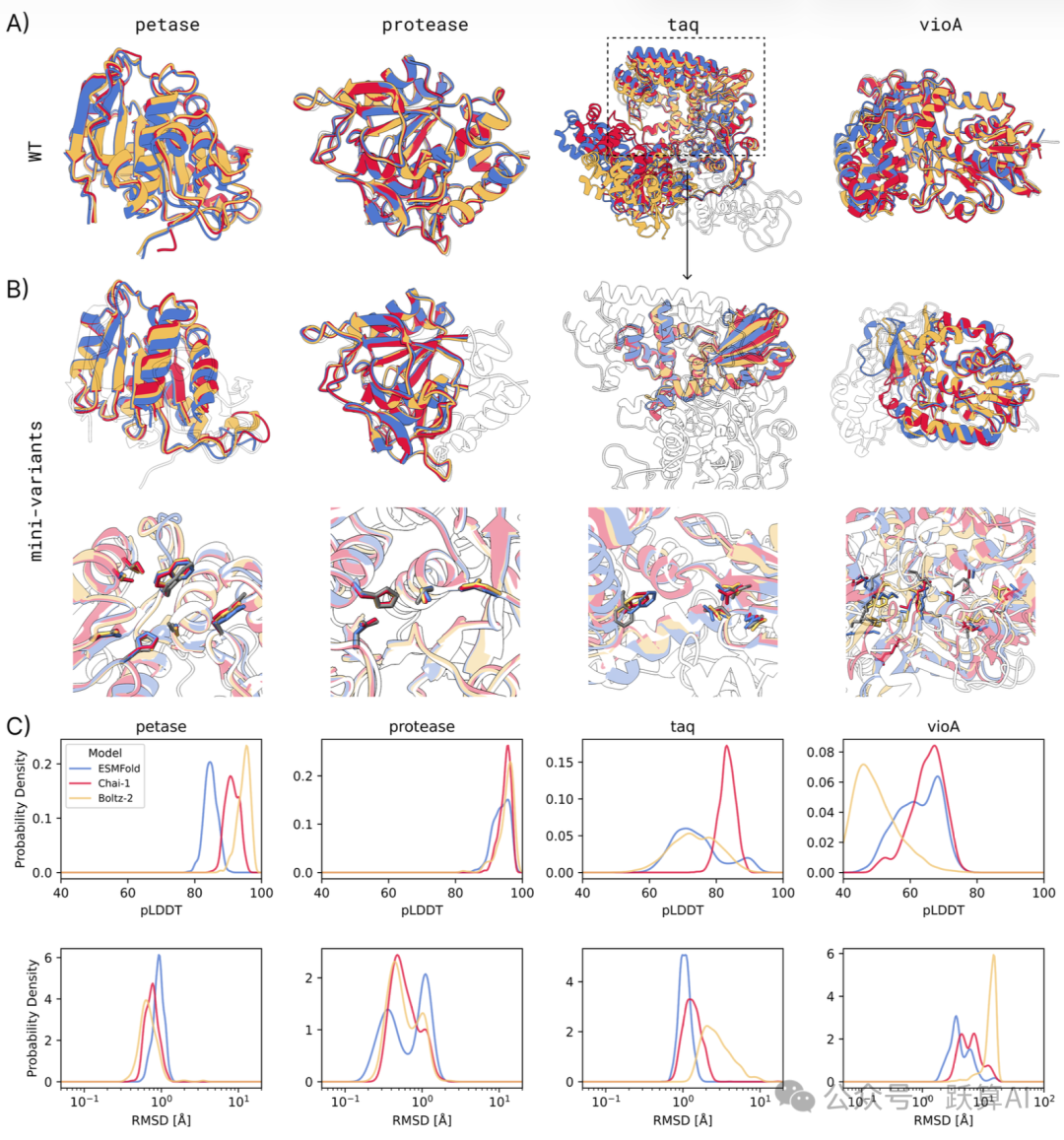
图 4 四种酶微型变体的多模型结构验证来源:论文 Figure 4
A:三种折叠模型对 WT 序列的预测结构叠合图 (黑色轮廓为晶体结构)。总体一致,taq 的两个结构域相对位置存在系统性偏差,这是模型本身的局限而非微型化造成的。
B (上):每种酶综合 RMSD 最低的 mini-variant 全局结构,与 WT 晶体结构 (黑色轮廓) 叠合。petase 和 protease 的全局折叠极度接近 WT;taq 出现局部旋转偏差;vioA 多个活性位点残基已明显偏移。
B (下):放大的关键催化残基 (C) 近景视图,直观展示活性口袋的保真程度。
C:各酶 mini-variants 的 pLDDT (上) 和 RMSD (下,对数坐标) 概率密度分布。ESMFold (蓝)、Chai-1 (红)、Boltz-2 (黄) 三条曲线重叠度越高,说明结构共识越强;petase 和 protease 三曲线几乎完全重合。
七、结果④:分子动力学 —— 动态视角下的活性位点稳定性
折叠算法是静态的,无法模拟热涨落对结构的影响。研究者对 top-16 候选分别进行 500 ns 全原子显式溶剂 MD 模拟 (OpenMM + AMBER14ff + TIP3P-FB),评估两个关键指标:
RMSF (均方根涨落):衡量每个残基在轨迹中的热运动幅度;
活性位点 RMSD 轨迹:以 WT 平均构型为参考,追踪活性位点重原子随时间的偏差。
最佳结果令人鼓舞:petase (mini-petase-138) 和 protease (mini-protease-35) 的活性位点 RMSD 全程维持在~1-2 Å 附近,与 WT 本身的热涨落幅度相当。taq 和 vioA 的部分变体也展现出可接受的稳定性,但整体方差更大。
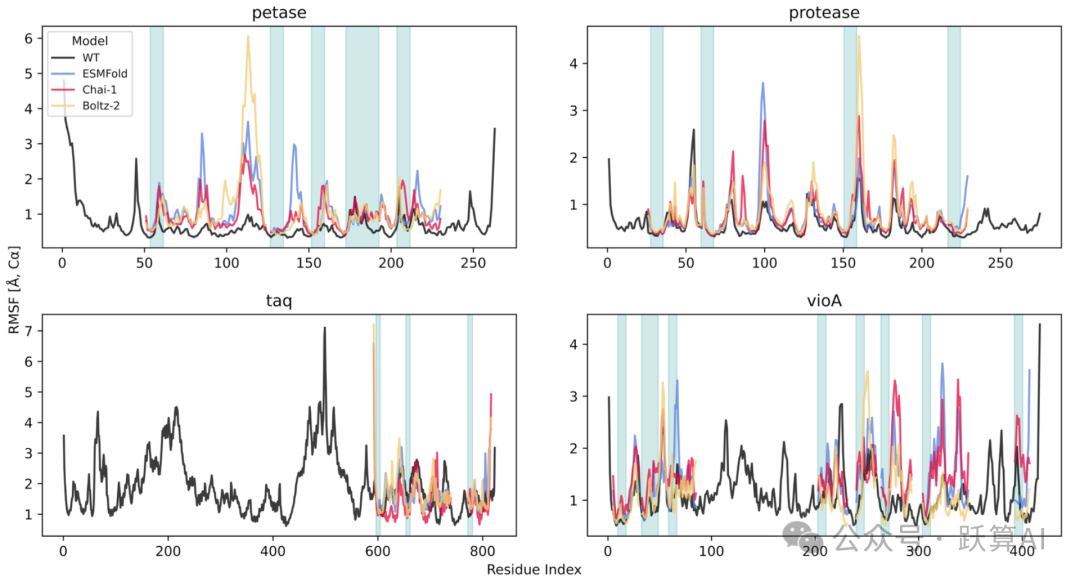
图 5 最佳微型化变体的分子动力学验证 (RMSF 图) 来源:论文 Figure 5
· 每个子图对应一种酶 (petase/protease/taq/vioA)。横轴为残基编号,纵轴为 Cα RMSF (Å)。
· 黑色曲线为 WT 参考;蓝 / 红 / 黄曲线分别为以 ESMFold/Chai-1/Boltz-2 折叠结构为初始构型的 MD 模拟结果;青绿色阴影区域标注不可变残基 (I) 位置。
· 核心观察:在不可变区域内,最佳 mini-variant 的 RMSF 曲线与 WT 大致吻合,尽管全局折叠已完全不同;petase 约 120 号残基附近出现新的高涨落峰,反映局部铰链在压缩后被放大。
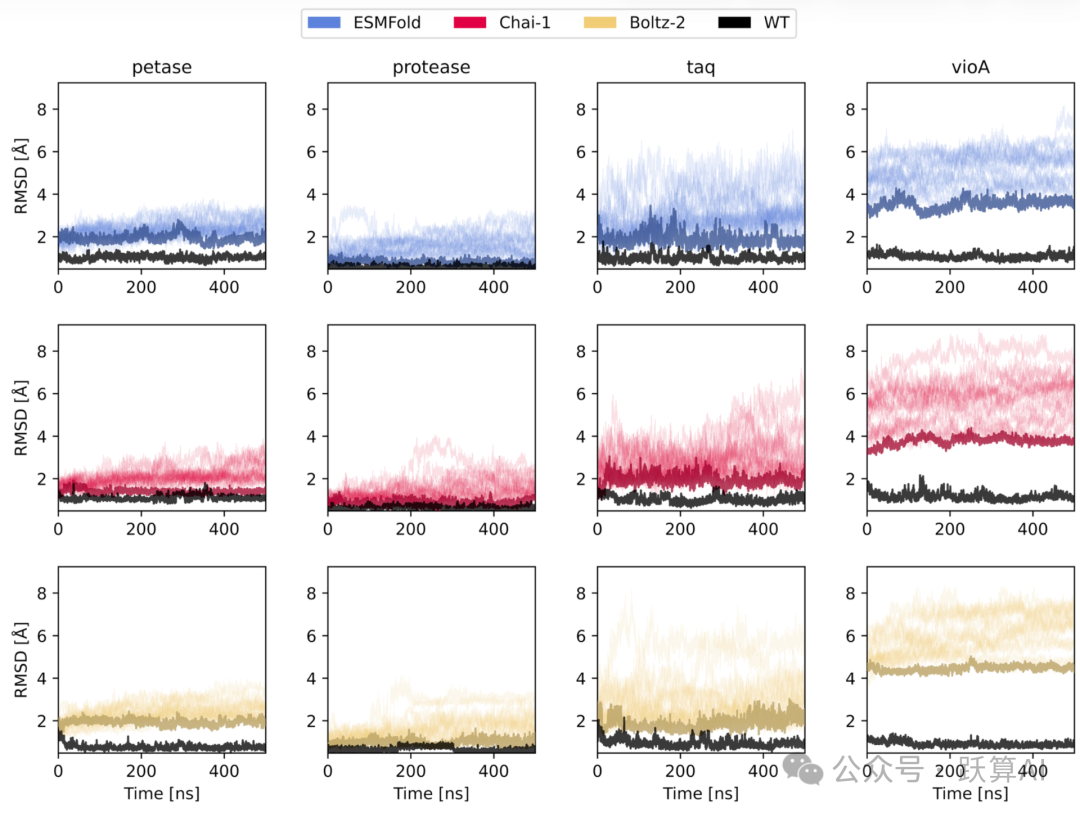
图 6 MD 轨迹中活性位点 RMSD 随时间的演化来源:论文 Figure 6
· 行:三种折叠模型 (ESMFold/Chai-1/Boltz-2);列:四种酶。每个面板中黑色为 WT,彩色细线为 16 条 mini-variant 轨迹,颜色最深的为 RMSD 均值最低的最优变体。
· 关键结论:petase 和 protease 的最优变体 RMSD 全程贴近 WT 水平 (1-2 Å);taq 和 vioA 部分变体有较大漂移,需进一步筛选。
八、讨论:这套方法能打吗?
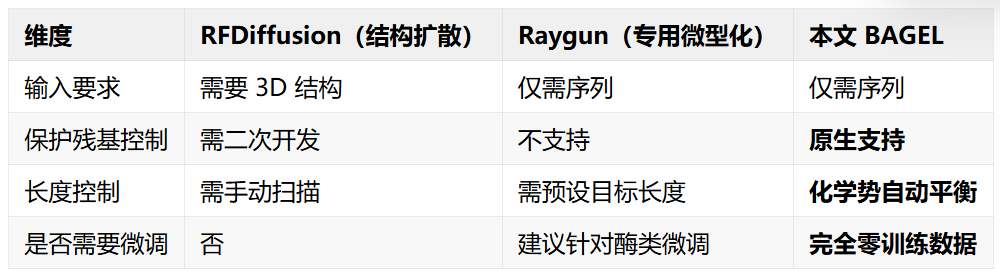
和竞品的对比
论文将该方法与两类主流方案对比:
四种酶的实际结果
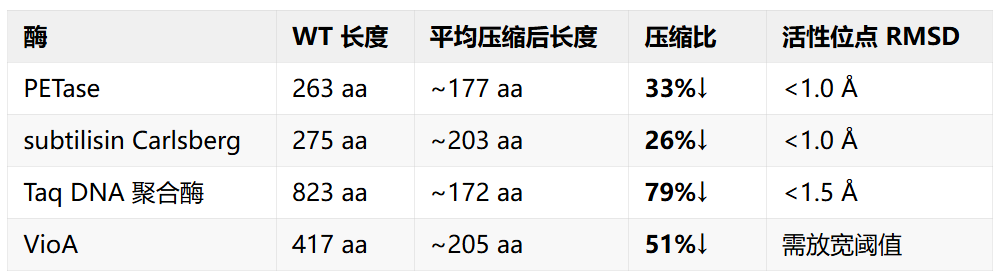
值得注意的是,Taq 的巨幅压缩 (79%) 实际上去掉了 N 端的 5' 核酸酶结构域,只保留 C 端聚合酶域 —— 这对只需要 DNA 聚合活性的应用可以接受,但用于探针法 qPCR 则不适用。
开放问题
作者们很坦诚地指出,计算层面的验证并不能保证实验成功:SolubleMPNN 打分系统性地低估了这些 de novo 序列 (可能只是训练集分布外效应,而非真的不可溶),MD 模拟也没有纳入辅因子 (如 VioA 的 FAD)。真正的答案,还需等待实验室的湿实验数据。
九、方法小结
输入:WT 酶序列 + 文献中已知的催化关键残基
Step 1 定义不可变区域 I (关键残基 + 左 4 右 3 缓冲)
Step 2 BAGEL MC 优化 (50 条 ×10 万步),同时允许替换 / 插入 / 删除
↳ 能量 = 嵌入相似度损失 + 序列长度惩罚
Step 3 ESMFold 折叠所有候选,按 RMSD + pLDDT 粗筛
Step 4 ESMFold / Chai-1 / Boltz-2 三模型共识精筛
+SolubleMPNN log-likelihood 排序 → Top-16
Step 5 500 ns 全原子 MD 模拟,评估活性位点动态稳定性
最终实验候选序列 (~few per enzyme)
输出:开源于 Zenodo 的全部候选序列
十、写在最后
这篇工作最吸引人的地方,不只是那一串漂亮的 RMSD 数字,而是方法论层面的简洁:不需要结构数据、不需要重新训练模型、不需要领域专用数据集,只需要一个预训练 PLM 和一个精心设计的能量函数,就能把「序列空间里的大海捞针」变成一道可以系统求解的优化问题。随着 PLM 和结构预测模型的不断迭代,BAGEL 框架也会「免费」跟着涨水涨船高。更令人期待的是后续实验验证的结果 —— 毕竟,硅基的预言终究要在碳基的世界里兑现。
所有候选序列已开源:Zenodo DOI: 10.5281/zenodo.18854113
原文信息: Lála J, Agrawal H, Dong F, Wells J, Angioletti-Uberti S.An Energy Landscape Approach to Miniaturizing Enzymes using Protein Language Model Embeddings.bioRxiv, March 4, 2026. DOI: 10.64898/2026.03.04.709378
文章转载自微信公众号:https://mp.weixin.qq.com/s/KZaAPBVttTFpnM2GuoQqEA?mpshare=1&scene=1&srcid=0311adSBCPjvHHBeZXjlYArQ&sharer_shareinfo=17110d12960b395f965fe33c95ea1c63&sharer_shareinfo_first=17110d12960b395f965fe33c95ea1c63&from=industrynews&color_scheme=light#wechat_redirect
原文链接:跃算AI | 





















