本帖最后由 薛定谔了么 于 2026-4-28 15:03 编辑
说实话,现在的3D分子生成领域简直卷出天际。
Diffusion已经被大家玩烂了,Flow Matching也渐渐让人审美疲劳。我们都在焦虑地观望:在这个后Diffusion时代,除了堆算力和加层数,数学上还有没有新活可整?难道只能坐等Transformer一统天下?
今天这篇ICLR 2025的DrugFlow,恰恰就是在这个时间节点上,给我们打了一针强心剂。这篇来自EPFL(Bruno Correia组) 和Oxford(Michael Bronstein组) 两大豪门的联手之作,并没有抛出一个全新的不知名名词,而是极其硬核地向我们展示了:当流匹配(Flow Matching)、马尔可夫桥(Markov Bridges)和黎曼流形(Riemannian Manifolds) 这三者合体时,会发生什么?
我们读这篇Paper,不是为了看它刷榜,而是为了搞清楚:这种"连续+离散+流形"的混合数学架构,会不会就是下一代生成模型的终极形态?所谓的"桥(Bridge)"到底是不是接棒Diffusion的下一个风口?
来,让我们带着"找茬"和"寻宝"的心态,拆解一下这篇集大成者。
1.开始之前:小白也能懂的"生成式桥梁学"(Generative Bridges 101)
在深度学习里,我们经常听到Diffusion(扩散)。普通的Diffusion就像是一个醉汉走路(Random Walk):
前向(Forward):醉汉从酒吧(真实数据分布)出发,越走越远,最后倒在路边草丛里(纯高斯噪声)。
反向(Reverse):我们训练一个神经网络(警察),试图把醉汉从草丛里一步步扶回酒吧。
那么,什么是"桥(Bridge)"呢?
1.1 什么是"桥"(The Bridge)?
通俗解释:普通的随机过程是"管杀不管埋",只管起点,终点听天由命。而"桥"是一种被强行约束了两端的随机过程。这就好比:醉汉从酒吧出来(t=0),但他老婆下了死命令,不管你中间怎么晃悠,t=1时必须准点出现在家门口。这种"起点固定、终点也固定"的路径,在数学上就叫"桥"。
专业定义:给定随机过程Xt,如果我们在已知X0=a的条件下,再附加一个条件 XT = b,那么在这个条件概率下的过程{Xt | X0=a, XT=b}就称为桥。
1.2 什么是扩散桥(Diffusion Bridge)?
通俗解释:我们在做图像生成时,通常是一端是噪声,一端是图像。普通的Diffusion是:我先假装把图像加噪变成噪声,再学着反悔。而Diffusion Bridge更像是一种"定向运输"。比如我想做"图像翻译"(把斑马变成马):
t = 0:斑马的分布
t = 1:马的分布
Diffusion Bridge:直接学习一种扩散过程,让像素点从斑马的状态,平滑地漂移到马的状态。它不再必须从中途的"纯噪声"过境,而是直接连接两个分布。
本质:它是求解两个概率分布之间变换的SDE(随机微分方程)解。
1.3 什么是Markov Bridge(本文用到的概念)?
Markov Bridge不完全等于Diffusion Bridge,但在广义上它们是一家人。
区别在于数据类型:
通常说的Diffusion/Score-based Bridge处理的是连续数据(Continuous),比如像素值、原子坐标(0.5, 1.2, -3.4)。即使加了噪声,它还是个连续的数值。
Markov Bridge(特指Discrete Bridge)处理的是离散数据(Discrete/Categorical),比如:这个原子是"碳"还是"氮"?这个键是"单键"还是"双键"?
通俗解释DrugFlow里的Markov Bridge:想象你要把单词"CAT"变成"DOG"。
在连续空间(Diffusion),你可以把C慢慢变模糊,像墨水晕开一样变成D。
在离散空间(Markov Bridge),你不能有"半个C"。原子类型的变化是跳跃的(Jump)。
Markov Bridge的做法:我在t=0时有一个起始状态(比如真实原子类型),在t=1时有一个目标状态。我在中间的时间点建立一座桥,计算在t时刻,这个原子从"碳"跳变为"氮"、"氧"或者保持不变的概率转移矩阵。
它的数学工具不是SDE(随机微分方程),而是Rate Matrix(速率矩阵)和Master Equation(主方程)。
关键区别:与Diffusion"从数据到噪声再回来"不同,Markov Bridge可以直接在两个有意义的离散状态之间建立桥梁,不需要经过"纯噪声"中转站。这也是DrugFlow能高效处理离散原子/键类型的数学基础。
2.引入与背景(Hook)
痛点场景化:现在的AIDD工程师在做SBDD(基于结构的药物设计)时,经常面临一种"精神分裂"的痛苦。
如果你用Diffusion做分子生成,原子坐标算得挺准,但生成的原子类型经常乱填(比如给碳连了5条腿)。
如果你用自回归模型(像写句子一样生成分子),化学效度高了,但3D结构往往扭曲得没眼看。
更别提,很多时候我们不仅要生成分子,还得微调蛋白口袋的侧链(Induced-fit),这时候现有的模型基本都要歇菜。
本文定位:DrugFlow是一篇"集大成者"。它不再试图用一种数学工具解决所有问题,而是承认了分子的复杂性——它既有连续的几何形状,又有离散的化学属性。于是,作者极其硬核地在一个模型里融合了三种不同的数学流派。这是一篇真正想解决"多模态物理约束"的落地级论文。
3.方法论的"去黑盒化"解读(The Logic)
别被公式吓到了,DrugFlow的核心逻辑其实非常直观。想象你在指挥一个交响乐团,弦乐(坐标)、管乐(原子类型)和打击乐(侧链角度)各有各的乐谱,但必须在你的指挥棒下完美同步。
Input / Output
输入:蛋白口袋结构(Cα坐标 + 氨基酸类型 + 残基内所有原子的相对位置向量)
输出:配体的3D坐标、原子类型、化学键类型,以及(划重点)口袋侧链的构象变化
核心直觉:Flow Matching + Markov Bridge
为了处理三种不同的数据类型,作者没有强行用一种算法套所有,而是搞了个"混合三打":
a.对于坐标(连续,):使用Euclidean Flow Matching(欧氏流匹配)。你可以把它理解为比Diffusion更直球的生成方式。Diffusion是醉汉走路,跌跌撞撞去噪;Flow Matching则是试图拉一条直线(或近似直线),直接把高斯噪声"平滑传输"到目标分子结构。
b.对于原子/键类型(离散,Categorical):使用Markov Bridge Model。这就好比我们在两个离散状态(比如训练数据中的真实原子类型分布)之间架了一座概率桥梁,让状态在时间轴上按照精心设计的转移概率演变。
c.对于侧链角度(周期性,Torus ):使用Riemannian Flow Matching。因为角度是360度循环的(环面/超环面),不能用普通的欧式距离,作者在流形上定义了测地线流匹配,专门用来预测蛋白侧链的角。
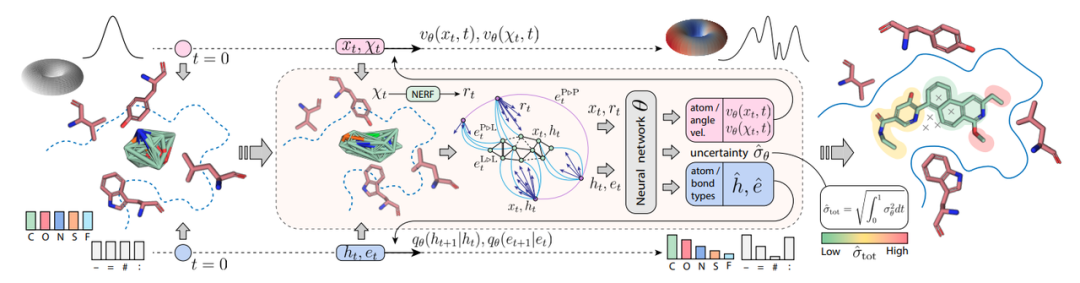
Figure 1:左边展示了输入的三种数据流:坐标(连续,从高斯先验出发)、类型(离散,从均匀或边缘分布先验出发)、角度(环面,从均匀先验出发)。中间的核心网络是一个等变的异构GNN(基于GVP),它同时输出速度场(Velocity,用于更新坐标和角度)和类型预测logits(用于计算Markov Bridge的转移概率)。右边展示了那个很聪明的"Virtual Node"机制。
两个让人眼前一亮的巧思
自带"裁剪刀"的Virtual Node
为了解决"原子数定死"的问题,作者引入了一个虚拟节点(Virtual Node)类型。模型在生成过程中,可以把某些原子标记为"虚拟的"(即不存在)。这就好比给了模型一把剪刀,它发现原子太多挤不进口袋时,就把多余的原子"剪掉"。虚拟节点的坐标被设定在配体质心位置,为网络提供了一个明确的回归目标。这让DrugFlow实现了端到端的尺寸自适应,无需额外的Size Predictor。
内生不确定性估计(Uncertainty Head)
作者给模型加了一个简单的回归头,预测当前步骤Flow Matching回归误差的方差\sigma^2。通过最大化似然函数推导出的损失函数(Eq. 1),不需要额外的OOD数据训练,这个方差自然就学会了识别模型"不确定"的区域。最终的不确定性分数通过沿采样轨迹积分得到(Eq. 2)。
网络架构:一个GNN如何同时吐出连续和离散输出?
到底什么样的神经网络能同时吐出"原子坐标(连续向量)"和"原子类型(离散概率)"?是不是搞了三个分开的网络?
并不是。 作者设计了一个非常精巧的Equivariant Heterogeneous GNN(等变异构图神经网络)作为共享骨干(Backbone)。
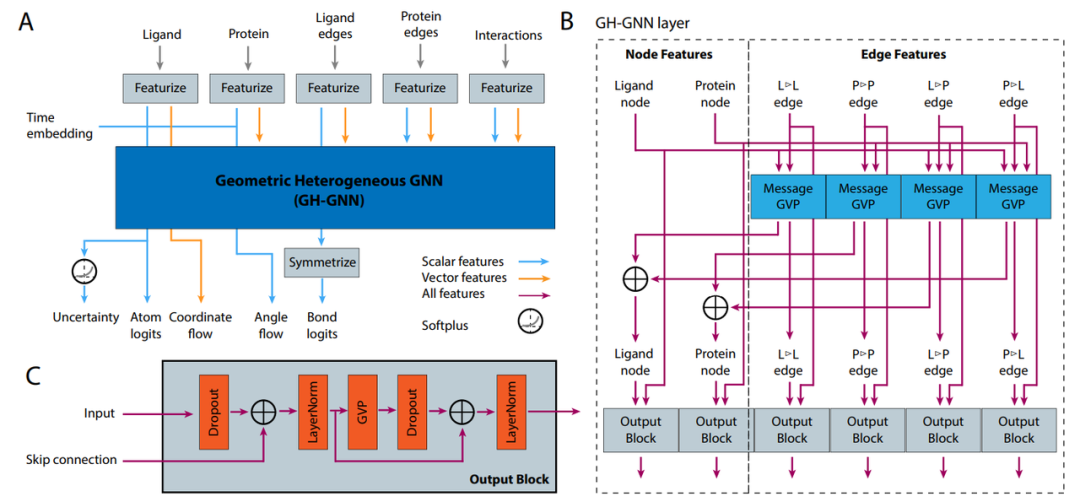
Figure 6:这张图展示了DrugFlow的"微观操作"。它不是普通的GNN,而是基于GVP (Geometric Vector Perceptrons) 思想的架构。
核心设计要点:
a.双流处理(Scalar & Vector):网络内部一直维护着两套数据流——标量特征h(如原子类型Embedding、时间步编码)和向量特征v(如坐标差\Delta x、原子到残基内其他原子的方向向量)。标量负责"化学属性",向量负责"几何结构"。
b.异构图设计(Heterogeneous):四种边类型(L▷L配体内、P▷P蛋白内、L▷P、P▷L)各有独立的消息传递权重。蛋白节点用残基级别表示(以Cα为坐标),但通过向量特征保留了全原子信息。
c.等变性保证:所有作用在向量上的操作都通过GVP实现,确保预测的速度场在全局旋转/平移下正确变换。
d.输出分流:同一个骨干网络最后分出多个输出头——坐标速度场vθ(xt, t)、角度速度场vθ(\chi_t, t)、原子类型logits^ht、键类型logits \hat{e}t,以及可选的不确定性\hat{\sigma}。
4.【掘金时刻】那些论文里没明说,但极具参考价值的Tricks
读代码和Appendix才能发现的工程细节,往往比正文更值钱。我为大家挖掘了以下3个"Model Engineering Highlights":
Trick 1:基于距离截断的图构建(Distance-Cutoff Graph)
DrugFlow的图构建策略值得关注:
配体原子间:完全连接(complete graph),因为任意两个原子之间都可能形成化学键
蛋白-配体间、蛋白-蛋白间:基于10Å距离截断动态构建边,平衡了计算效率和信息传递范围
这种设计让模型在每个采样步骤都能根据当前原子位置更新蛋白-配体的交互图,捕捉到随着生成过程逐渐形成的局部化学环境。
Trick 2:虚拟节点的"幽灵状态"(The Virtual Node Trick)
关于原子数自适应,文中有一个细节处理很棒:
操作:设定一个最大虚拟节点数N{max}(论文中=10)。训练时随机添加n{virt} \sim U(0, N_{max})个虚拟节点。
坐标设定:虚拟节点的坐标放在配体质心位置,为网络提供明确的回归目标。
类型演变:Markov Bridge不仅学习碳、氮、氧之间的跳变,还学习"真实原子"与"虚拟(不存在)"之间的跳变。
推理时:如果采样结束时某个节点的类型是Virtual,直接丢弃。
这把"尺寸预测"内化为"类型预测"的一部分,非常优雅。但注意:N_{max}=10意味着模型最多只能自适应调整约±5个原子,对于需要生成大分子(如PPI抑制剂)的场景可能不够用。
Trick 3:黎曼流形上的多项式调度器(Polynomial Scheduler for Torus)
对于侧链角度的流匹配,作者使用了一个多项式调度器\kappa(t) = (1-t)^k(k=3),控制测地线距离的收缩速率:
相比指数调度器\kappa(t) = e^{-ct},多项式调度器严格满足边界条件\kappa(0)=1, \kappa(1)=0,理论上更为严谨。这个小改动对采样质量有显著提升。
5.实验结果的"侦探式"分析(The Evidence)
作者在CrossDocked数据集上进行了测试(100K训练对,100个测试蛋白)。比起那些只盯着Vina Score看的论文,这篇论文的Evaluation显得格外"清流"——它核心关注的是分布学习能力,而非绝对指标刷榜。
作者的核心观点:生成模型的目标是学习数据分布,而非超越数据分布。没有额外微调或采样策略的情况下,期望模型大幅超越训练集的指标是不合理的。
证据一:不仅要分高,还要"像样"
作者大量使用了Wasserstein距离和Jensen-Shannon散度来衡量生成分子的属性分布与真实药物分布的差异。
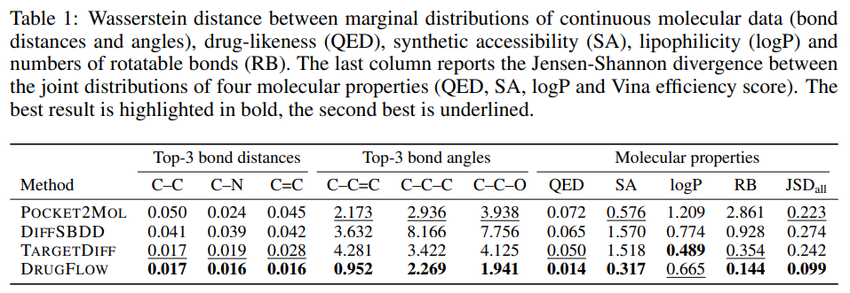
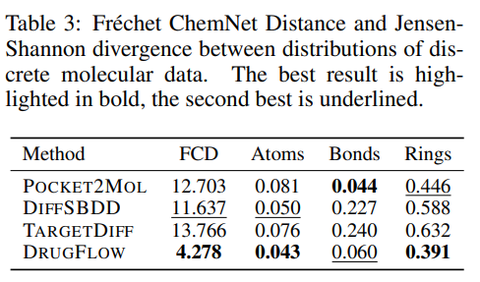
Table 1 & 3:JSD_all是QED、SA、logP、Vina efficiency四个属性联合分布的JS散度。
结果显示,DrugFlow生成的分子的键长、键角分布,几乎完美复刻了真实分子。这听起来很简单?其实很难!很多Diffusion模型生成的分子虽然对接分高,但一看结构,各种扭曲的键长,根本造不出来。DrugFlow在Fréchet ChemNet Distance(FCD)上也以4.28的成绩完爆其他方法(12-14),说明它覆盖了更多化学空间的modes。
证据二:不确定性 = 避雷针
这是我觉得最性感的一个实验结果。
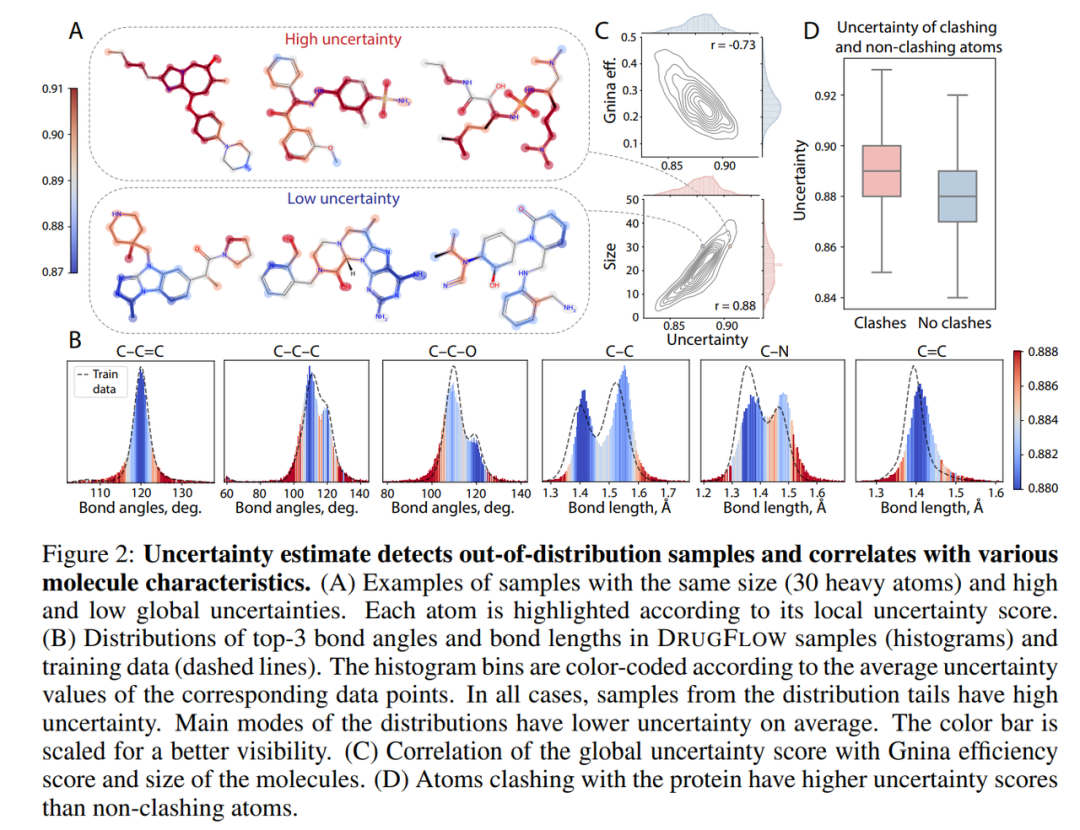
Figure 2解读:
Panel A:同样大小(30个重原子)的分子,高/低不确定性样本的可视化。每个原子按其局部不确定性着色。
Panel B:键长/键角分布的直方图,颜色编码表示平均不确定性。分布尾部(异常值)的不确定性显著更高,说明模型"知道"这些样本不太靠谱。
Panel C:全局不确定性与Gnina efficiency(r=-0.73)和分子大小(r=0.88)的相关性。
Panel D:发生碰撞(Clash)的原子具有显著更高的不确定性。这意味着模型自己其实"知道"这个原子放这儿不对劲。
工业价值:我们可以直接用这个不确定性来过滤掉那些不靠谱的生成结果,而不用去跑昂贵的MD或FEP。但需要注意,这个不确定性估计的是Flow Matching预测误差的方差,它能反映模型的"熟悉程度",但不能替代真正的binding affinity不确定性评估。
证据三:让蛋白"动"起来(FlexFlow)
作者展示了扩展版本FlexFlow的能力。在给定配体结构固定的条件下,FlexFlow能把原本relaxed的侧链重新采样到结合态的构象,RMSD中位数只有1.75Å(相比relaxed apo的1.98Å有显著提升)。

Figure 4解读:
Panel A:三种情况下的侧链RMSD分布——随机先验(很差)、relaxed apo(1.98Å)、FlexFlow生成(1.75Å)。
Panel B:三种大体积氨基酸(Arg, Lys, Trp)的\chi_1-\chi_2角度分布。FlexFlow样本(黑线)与训练集晶体结构(蓝色等高线)高度一致,正确捕捉了rotamer modes。
这证明了模型真的学到了一定程度的"诱导契合"物理规律,而不是死记硬背。
证据四:偏好对齐(Preference Alignment)
作者还顺手把LLM里的DPO(Direct Preference Optimization) 搬了过来,用来优化QED、SA、Vina efficiency和REOS过滤通过率。
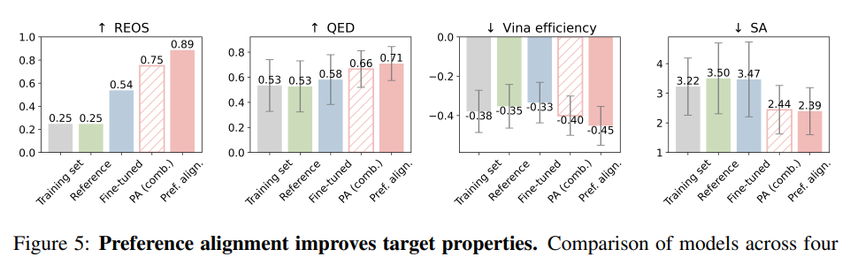
Figure 5解读:偏好对齐(红色)在所有四个指标上都超越了简单的Fine-tuning(蓝色)和基准模型(绿色)。组合偏好对齐(虚线)也表现出色。
关键创新点是:作者同时在Flow Matching(连续)和Markov Bridge(离散)两个框架上应用了DPO,实现了真正的多域偏好对齐(MDPA)。不需要复杂的强化学习,直接用对比数据微调,效果拔群。
6.点评:优势、局限与避坑指南(The Verdict)
优势(Pros)
物理直觉极佳:通过"多域分布学习",DrugFlow不仅仅是在生成图,而是在生成符合物理约束的3D实体。键长、键角分布的高保真度是最好的证明。
评价范式革新:作者力推"分布学习质量"而非"绝对指标刷榜"的评价方式,这对整个领域都有指导意义。用Wasserstein距离和FCD评价生成模型,比直接比Vina Score更科学。
工程化细节满分:Virtual Node、Uncertainty Head、MDPA等设计都非常实用,代码开源(GitHub),复现友好。
FlexFlow开创性:虽然只是侧链柔性,但这是SBDD生成模型首次认真处理蛋白构象采样问题。
局限与槽点(Cons & Risks)
a."虚拟节点"的上限:虽然Virtual Node很聪明,但在训练时,作者只设置了N{max}=10。这意味着模型最多只能自适应调整约±5个原子。如果你初始给的原子数严重过剩(比如给个100原子的框去填一个小口袋),模型还是会两眼一抓瞎(见Figure 3B)。对于PPI抑制剂等大分子场景,可能需要增大N{max}。
b.骨架还是刚性的:FlexFlow虽然让侧链动了,但蛋白Backbone(主链)还是死的。在处理那些Loop区大尺度运动的靶点(比如激酶的DFG-in/out切换、GPCR的TM螺旋运动)时,这个模型依然无能为力。
c.FlexFlow的条件依赖:Figure 4A的实验是固定配体采样侧链,而非真正的配体-蛋白协同采样。当配体骨架与训练集差异很大时,侧链响应可能不准确。
d.没有实验验证:所有评价都是计算指标,没有prospective的binding assay或cell assay验证。这是整个SBDD领域的通病,但作为"落地级论文"的定位,缺少实验数据是一个遗憾。
e.数据集偏差问题:CrossDocked本身存在cross-docking artifacts,很多pose质量堪忧。虽然作者用PoseBusters过滤了训练集,但"垃圾进垃圾出"的风险依然存在。不确定性估计的意义之一,正是帮助识别这些问题区域。
工业界落地建议
别拿来做大规模虚拟筛选:杀鸡焉用牛刀,算力烧不起。DrugFlow每生成一个分子需要500步采样,比autoregressive方法慢很多。
最适合场景:Hit-to-Lead阶段的骨架跃迁(Scaffold Hopping)。当你有一个大致的结合模式,想要保持关键相互作用但换个骨架,并且希望能处理口袋微小的侧链调整时,DrugFlow/FlexFlow是可以尝试的工具之一。
利用Uncertainty:一定要把那个不确定性输出利用起来,作为一个免费的粗筛Filter,可以帮你省下大量后续对接和FEP的计算资源。但记住,它不能替代binding affinity的精确计算。
考虑偏好对齐:如果你有明确的优化目标(比如提高SA、降低毒性alerts),MDPA提供了一个比RL更稳定的微调方案。
7.思考与未来(Takeaway)
DrugFlow告诉我们,未来的AIDD模型一定不是"黑盒"的暴力美学,而是"物理几何 + 深度学习"的精细化融合。
三个值得关注的趋势:
a.多域生成成为标配:分子本身就是连续(坐标)+离散(类型)+周期性(角度)的混合体,未来的模型必须能原生处理这种混合数据。
b.Bridge > Diffusion?:Schrödinger Bridge / Markov Bridge这些听起来高大上的数学名词,正在快速进入落地阶段。相比Diffusion"必须经过纯噪声"的设定,Bridge的"两端都有意义"可能是更自然的建模方式。作为算法工程师,现在去补一补随机过程和最优传输(Optimal Transport) 的课,绝对是保值增值的最佳投资。
c.蛋白柔性不可回避:FlexFlow只是开始。未来的SBDD模型必须能处理Backbone运动、Loop sampling、甚至allostery。这需要和蛋白结构预测/动力学模拟领域更深度的交叉。
8.深度Q&A
Q1: DrugFlow号称"分布学习器",但它真的学到了蛋白-配体相互作用的物理本质吗?还是只是一个高级的数据过拟合器?
部分学到了,但有限。
支持"学到物理"的证据:clash原子的不确定性显著更高(Figure 2D),说明模型学会了避免空间碰撞;键长键角分布高度还原真实分子。
但更诚实的判断是:DrugFlow更像一个高保真的数据分布复制器。它学到的是"训练集里的分子长什么样",而非"为什么这样的分子能结合"。真正的物理理解需要prospective实验验证——目前一个都没有。
启发:评价生成模型时,"分布匹配得好"≠"理解了底层物理"。前者是统计相关,后者需要因果验证。
Q2: 不确定性估计听起来很美,但它估计的是Flow Matching回归误差,这和"药物设计的不确定性"是一回事吗?
不是一回事,但有交集。
DrugFlow的不确定性反映的是:模型对当前输入的"熟悉程度"(认识论不确定性)。它能告诉你"这个区域我没见过",但不能告诉你"这个分子的binding affinity误差是多少"。
真正的药物设计不确定性来源更广:力场精度、熵效应、溶剂化、蛋白柔性、off-target……这些远超flow matching误差能覆盖的范围。
实用建议:把它当粗筛过滤器用,剔除明显不靠谱的样本;但不能替代FEP/MD等下游验证。
Q3: FlexFlow让侧链"动"起来了,但这是真正的induced-fit吗?当配体骨架完全改变时,侧链响应还准吗?
是受限的induced-fit,不是通用的。
三个关键限制:
1.Backbone是死的——Loop运动、DFG翻转等大尺度变化无法处理
2.条件是单向的——实验是"固定配体采样侧链",而非配体-蛋白协同调整
3.泛化存疑——训练数据来自holo结构,面对全新骨架的配体时,学到的响应模式可能不适用
适用场景:配体骨架与训练集相似、侧链调整幅度小(<2Å)、不涉及cryptic pocket opening。
不适用场景:全新化学空间、需要诱导loop运动、变构位点设计。
Q4: 为什么选Flow Matching而不是Diffusion?除了"更火"之外,有本质优势吗?
有,但没有宣传的那么大。
Flow Matching的真正优势:
轨迹更直:学习近似线性的传输路径,采样效率更高
训练更稳:直接回归向量场,不涉及score estimation的数值问题
理论更美:与最优传输自然衔接,有更多数学工具可用
但对于SBDD这个具体任务,Flow Matching vs Diffusion的性能差异没有想象中大。真正的提升来自:多域联合建模、Virtual Node、Uncertainty Head这些工程设计。
启发:别被数学框架的"高级感"迷惑,工程细节往往比理论选择更重要。
Q5: 所有评价都是计算指标,没有一个prospective实验验证。这篇论文的结论可信吗?
计算结论可信,药物设计价值存疑。
论文的计算实验设计是扎实的:Bootstrap统计检验、多维度分布比较、消融实验齐全。在"生成分子的统计特性是否接近训练集"这个问题上,结论是可信的。
但"统计特性接近"≠"能设计出好药"。整个SBDD生成模型领域都面临同一个尴尬:用Vina评价Vina-like的分子,本质是循环论证。
真正需要的验证:
至少一个靶点的prospective设计 + binding assay
与传统CADD方法(如SBVS+对接)的头对头比较
在全新蛋白家族上的泛化测试
结论:DrugFlow是目前最好的SBDD分布学习器之一,但它能否真正加速药物发现,jury is still out。
参考资料
Schneuing et al., "Multi-domain Distribution Learning for De Novo Drug Design", ICLR 2025.
代码:https://github.com/LPDI-EPFL/DrugFlow
文章改编转载自微信公众号:陷入鞍点
原文链接:https://mp.weixin.qq.com/s/8EahvDUe-tw2BYFgO28W2Q | 





















