本帖最后由 宇宙微尘 于 2026-5-18 23:15 编辑
本文基于《Nature Reviews Physics》发表的《Learning density functionals with differentiable DFT》,系统介绍了可微分密度泛函理论(Differentiable DFT)的技术框架与研究进展。该方法突破了传统 DFT 依赖手工设计交换关联泛函的瓶颈,通过将自洽场(SCF)计算流程全微分,实现了以数据驱动的方式学习泛函形式,在保留物理约束的同时,为平衡模拟精度与计算效率提供了全新路径,有望解决长程相互作用等传统方法难以处理的难题,推动材料科学与量子化学的范式革新。

当我们谈论锂电池的能量密度、新型催化剂的反应效率,或是抗癌药物与靶点蛋白的结合能力时,这些看似遥远的应用背后,都依赖着同一套核心工具 —— 密度泛函理论(Density Functional Theory, DFT)。作为量子化学与材料科学领域的 “主力军”,DFT 自上世纪 60 年代诞生以来,就承担着从微观电子结构出发,预测物质宏观性质的重任。从硅基半导体的能带结构,到二维材料的力学性能,再到分子晶体的堆积方式,如今超过 80% 的材料模拟研究都以 DFT 为基础。但鲜为人知的是,支撑这套工具的核心,却是一个始终无法完美解决的 “近似难题”—— 交换关联泛函。
一、传统 DFT 的 “天花板”:手工泛函的百年困局
DFT 的理论基石是霍恩伯格 - 科恩定理,它证明了多电子体系的所有基态性质,都可以由电子密度唯一确定。基于这一定理,我们可以将体系的总能量拆分为多个部分:
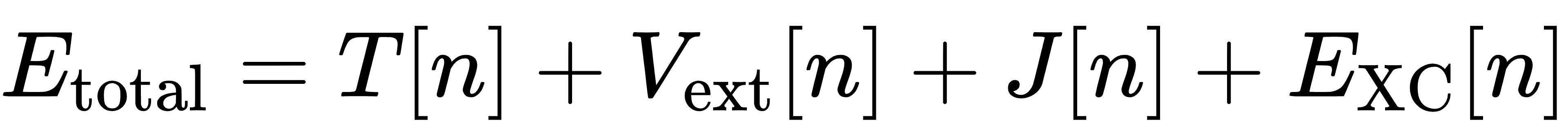
其中,T[n]是电子的动能,Vext[n]是外势场(比如原子核带来的库仑势),J[n]是电子之间的经典库仑排斥作用,而EXC[n]就是那个决定了 DFT 精度上限的交换关联泛函。它描述了电子之间的量子力学相互作用,包括泡利不相容原理带来的交换效应,以及电子运动关联带来的相关效应。
问题的关键在于,我们无法直接写出EXC[n]的精确表达式,只能通过近似来构建它。在过去的半个多世纪里,科学家们就像 “盲人摸象” 一样,通过物理直觉、数学推导和经验数据,手工设计了一代又一代的泛函模型:
● 从最早的局域密度近似(LDA),到考虑密度梯度的广义梯度近似(GGA),这些模型计算效率高,但在处理分子间作用力、强关联体系时误差较大;
● 再到引入部分精确交换能的杂化泛函,虽然精度有所提升,但计算成本会呈指数级增长,处理中等规模的分子体系都需要超算运行数天。
更棘手的是,这些手工泛函往往存在 “水土不服” 的问题。一个在分子体系中表现良好的泛函,放到固体材料中可能误差陡增;而针对特定体系优化的泛函,又难以推广到其他场景。这种 “顾此失彼” 的困境,让 DFT 的发展陷入了 “精度与效率不可兼得” 的僵局,也成为了制约材料科学研究突破的重要瓶颈。
二、破局之道:可微分 DFT 如何让 AI 自己 “学会” 泛函?
可微分 DFT(Differentiable DFT)的出现,为这一困局带来了全新的解决方案。它的核心思路,是把整个 DFT 计算流程,从 “手工设计的固定算法”,改造为 “可训练的机器学习模型”,让 AI 从数据中自动学习更精准的交换关联泛函。
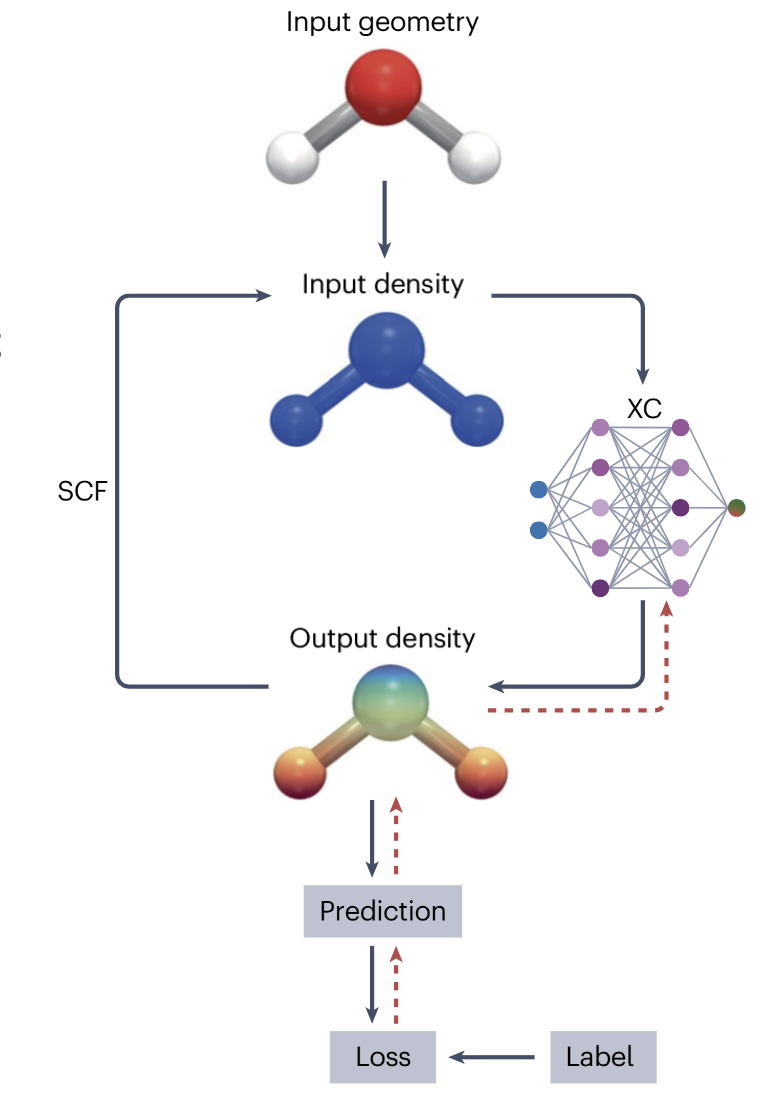
可微分DFT工作流程图
要理解这一突破,我们需要先拆解可微分 DFT 的核心创新:
1. 把交换关联泛函变成 “可训练的参数模型”
在传统 DFT 中,EXC[n]是一个固定的近似函数,比如 GGA 泛函的形式是预先设定好的,无法根据数据调整。而在可微分 DFT 中,我们将其替换为一个带参数的模型(比如神经网络):
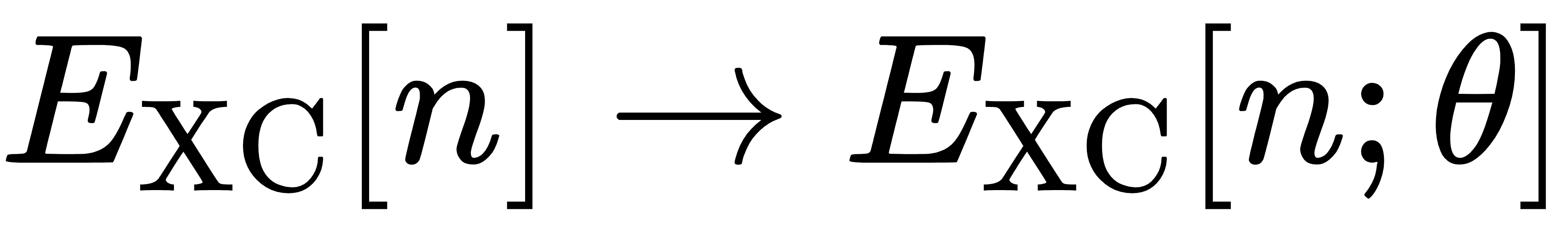
其中,θ是模型的可学习参数。我们的目标,就是通过训练数据来优化这些参数,让模型输出的交换关联能尽可能接近真实值。
2. 让整个自洽场(SCF)过程 “可微分”
DFT 的计算过程本质上是一个迭代求解的过程:先假设一个初始电子密度,求解单电子薛定谔方程得到轨道,再用轨道更新电子密度,重复这一过程直到密度收敛,这就是自洽场(SCF)迭代。在传统 DFT 中,这个迭代过程是 “黑箱” 式的,我们无法将误差信号反向传播到泛函参数上。
而可微分 DFT 的关键突破,就是让整个 SCF 迭代过程变得可微分。也就是说,当我们定义了一个损失函数,用来衡量模型预测结果与真实数据的差距:
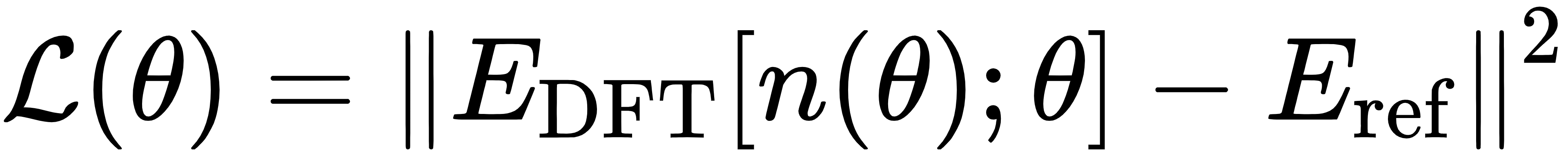
我们可以通过反向传播算法,计算损失函数对泛函参数θ的梯度,再用梯度下降算法直接优化参数。这里的n(θ)表示电子密度本身也是参数θ的函数,而整个从参数到密度再到能量的计算路径,都是可导的。
这就像给传统的 DFT 计算装上了 “反馈系统”,模型可以根据自己的预测误差,自动调整泛函的形式,而不用再依赖科学家的经验和直觉。
三、不止是 “精度更高”:可微分 DFT 的多重优势
可微分 DFT 带来的改变,远不止 “让 AI 设计泛函” 这么简单,它从根本上重构了泛函开发的范式,带来了多重突破:
1. 物理约束与数据驱动的完美结合
很多人担心,用机器学习训练出来的泛函会不会 “脱离物理常识”?可微分 DFT 很好地解决了这个问题。在训练过程中,我们可以将已知的物理规律编码到模型中,比如电子密度的归一性、交换关联能的对称性约束、热力学极限下的行为等。这些物理约束就像 “护栏”,确保模型学到的泛函不会违背基本物理规律,同时又能从数据中捕捉手工模型难以描述的复杂相互作用。
比如,在处理范德华力这类弱相互作用时,传统 GGA 泛函往往会严重低估相互作用能,而可微分 DFT 训练出的模型,可以直接从高精度的量子化学数据中学习到长程相互作用的特征,在保持计算效率的同时,大幅提升预测精度。
2. 为特定场景定制 “专家泛函”
不同的应用场景对泛函的要求截然不同:计算催化剂的反应路径,需要泛函能准确描述过渡态的电子结构;预测电池材料的电压,需要泛函能正确处理离子晶体中的电荷转移;而设计有机光电材料,则需要泛函能描述激发态的电子行为。
传统泛函往往是 “通用型” 的,难以同时满足所有场景的需求。而可微分 DFT 允许我们针对特定的体系或任务,用对应的数据集训练 “定制化泛函”。比如,我们可以用大量的有机分子数据集,训练一个专门用于有机光电材料的泛函;或者用金属氧化物的数据集,优化一个适合催化体系的泛函。这种 “场景化定制” 的思路,能让泛函的性能在特定领域发挥到极致。
3. 为长程相互作用难题提供新思路
传统泛函的一大短板,就是难以处理长程交换关联作用。局域和半局域泛函只依赖于某一点附近的电子密度,无法捕捉 3D 体系中长程电子相互作用;而杂化泛函虽然引入了部分精确交换能,但计算成本极高,难以应用到大规模体系中。
可微分 DFT 则为这一难题提供了折中方案。通过基于图的机器学习模型、注意力机制等技术,训练出的泛函可以在半局域的框架下,隐式地学习到长程相互作用的特征,既保持了接近传统 GGA 的计算效率,又能显著提升长程相互作用的描述精度。这意味着,未来我们可能用更低的计算成本,模拟更大规模的分子和材料体系,比如复杂的生物分子、多孔材料的气体吸附过程等。
四、挑战与展望:从 “实验室” 走向 “实用化”
当然,可微分 DFT 的发展也并非一帆风顺,目前仍面临着不少挑战:
首先,将整个 SCF 迭代过程做成可微分的,会显著增加计算复杂度。在迭代过程中,梯度可能会出现不稳定的问题,比如梯度爆炸或消失,需要引入正则化、稳定求解器等技术来控制误差。其次,训练出的泛函的通用性也是一个难题,如何让在特定数据集上训练的模型,能推广到从未见过的体系中,仍是当前研究的重点。此外,高质量训练数据的获取也存在门槛,高精度的量子化学计算或实验数据的成本较高,限制了大规模数据集的构建。
但这些挑战并没有阻挡科学家们探索的脚步。近年来,越来越多的研究团队开始尝试用可微分 DFT 解决实际问题:有的团队用它训练出了能准确描述水团簇相互作用的泛函,大幅提升了水的模拟精度;有的团队用它优化了催化剂的反应能垒预测,与实验结果的吻合度显著提升;还有团队将可微分 DFT 与分子动力学模拟结合,实现了更长时间尺度的材料模拟。
随着机器学习技术的进步和计算硬件的发展,可微分 DFT 正从 “概念验证” 阶段走向实用化。它不仅为 DFT 的发展注入了新的活力,更提供了一种 “用微分编程解决物理问题” 的新思路 —— 将传统的物理计算流程转化为可训练的模型,用数据驱动的方式优化近似模型,在保留物理约束的同时,突破传统方法的瓶颈。
可以说,可微分 DFT 正在叩开材料模拟的 “黑箱”。当 AI 开始学会描述电子之间的复杂相互作用,我们或许能打破传统泛函的限制,训练出既高效又通用的 “全能泛函”,让材料科学和量子化学的研究速度再上一个台阶。从新型锂电池材料的研发,到高效催化剂的设计,再到抗癌药物的筛选,这些应用场景都将受益于更精准、更高效的 DFT 模拟。而这一切的起点,正是可微分 DFT 带来的这场 “泛函革命”。
论文链接:https://www.nature.com/articles/s42254-026-00948-3 | 





















