当下Perturb-seq数据爆发期
从扰动数据出发的可解释、可生成的GRN推断框架的空白有待填补
Replogle等人2022年发布的K562全基因组规模数据集,包含了近万个基因的CRISPRi扰动和近200万个单细胞,数据已然是爆炸式增长了,但是我们用来从这些数据中推断基因调控网络(GRN)的计算方法,还停留在“扰动前时代”。GENIE3、GRNBoost2、PIDC这些主流方法本质上都是在计算基因对之间的统计关联,但没有一个全局的网络模型来整合扰动信息。这会导致遗漏在正常条件下被其他抑制信号掩盖、只有在特定扰动条件下才会显现的调控关系。
Caltech的Matt Thomson团队在Cell上发布的D-SPIN(Dimension-Scalable Single-cell Perturbation Integration Network)模型试图用一个不同的思路去解决这个问题,不是去统计基因对的关联,而是把统计物理的Ising模型“移植”进来,用能量函数去描述基因程序之间的相互作用,用条件依赖的外场建模扰动如何改变网络状态。这样模型不只是推断网络,它还能生成细胞状态分布,能模拟未观测的扰动组合,能绘制药物剂量组合的“相图”。

用物理学的能量景观
描述细胞状态
D-SPIN的数学核心是自统计物理的Ising模型(也叫自旋网络或马尔可夫随机场)。每个基因程序被离散化为三个状态(下调/-1、基线/0、上调/+1),基因程序之间的调控关系编码在一个交互矩阵J中(J>0表示激活,J<0表示抑制),而每个扰动条件则通过一个外场向量h来选择性地激活或抑制特定节点。整个细胞群体的转录状态分布由能量函数决定,其物理含义也非常直观,细胞群体像一组粒子在能量景观中活动,扰动像一个外力“倾斜”这个景观,让细胞群体流向新的状态谷。D-SPIN的创新是把学习问题分解为两部,首先是从所有扰动条件中联合推断一个统一的调控网络J,其次是推断每个扰动如何与这个统一网络交互(外场向量h),这样D-SPIN就可以把数千个扰动条件的信息“熔炼”到同一个网络模型中。
D-SPIN提供两个层级的架构:
程序级网络(Program-level)
用正交非负矩阵分解(oNMF)把数千个基因压缩为~30个共调控基因程序,然后用最大似然估计推断程序间的无向网络。这个网络是生成式的——可以模拟细胞状态分布,跟实际数据的余弦相似度超过90%。
基因级网络(Gene-level)
针对转录因子、激酶、磷酸酶等调控基因,用伪似然估计推断有向网络。这个算法可以扩展到数千个基因和数百万个细胞——256K细胞只需6小时(2个CPU核),而GENIE3等方法一周都跑不完。
为此,D-SPIN还设计了一个“调控因子发现”算法,通过从基因级网络中统计识别控制每个基因程序响应的关键调控因子,从而把“电路图”和“信号流”对接起来。
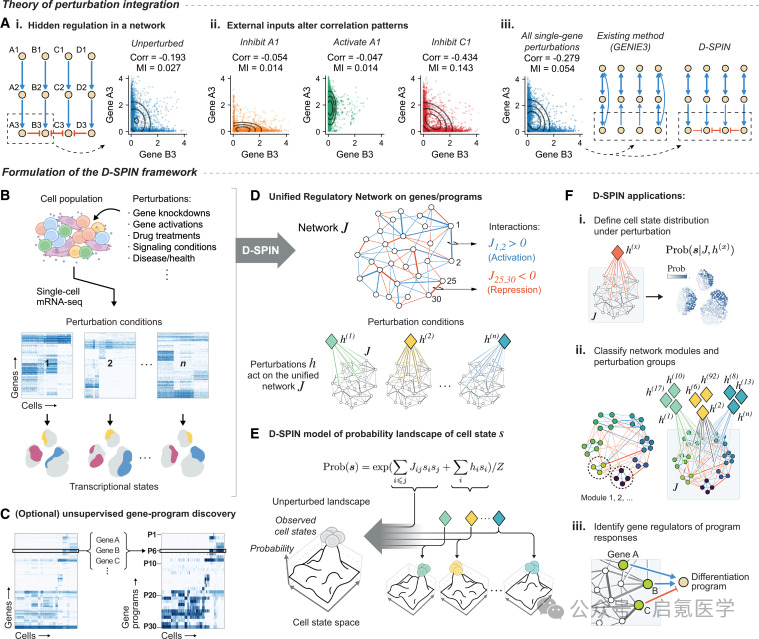
图1 D-SPIN框架概览:(A)扰动如何揭示隐藏的调控关系;(B-E)从单细胞mRNA-seq到统一调控网络的建模流程;(F)三大应用:细胞状态分布模拟、网络模块分类、调控因子识别
从11个节点到1000个基因
D-SPIN在合成数据和大规模网络上的基准测试中全面碾压现有方法,这种表现的核心原因就是其他方法无法区分基因表达变化是由内部调控还是外部扰动引起的,这种根本性的信息损失随着数据规模增大反而会让准确率先下降再缓慢回升,但D-SPIN则是扰动数量越多、推断越准。
BEELINE框架的造血干细胞(HSC)网络
D-SPIN的top-10边准确率达0.96(PIDC/GRNBoost2/GENIE3为0.77-0.83),AUPRC 0.87 vs 0.72-0.82。有向网络推断AUPRC达0.77 vs 其他方法的0.47-0.57。扩到1000个基因时,D-SPIN在模块化网络中保持0.913、随机网络0.773、无标度网络0.721,对应的竞争方法只有~0.5。
K562基因组规模Perturb-seq真实数据
D-SPIN推断的有向TF-靶基因网络与ChIP-seq数据库的对应率达11-15倍高于随机期望。对比差异表达(DE)分析,DE只找到了GATA1一个红系命运调控因子,D-SPIN同时识别出KLF1、NFE2、GFI1B、GATA1四个;DE没找到任何髓系调控因子,D-SPIN找到了SPI1和MEF2C。甚至,D-SPIN还唯一识别出了纯基于TF结合motif的方法从原理上就找不到的NPM1——一个通过BCR-ABL1激酶介导的转录后机制同时抑制两种细胞命运的基因。
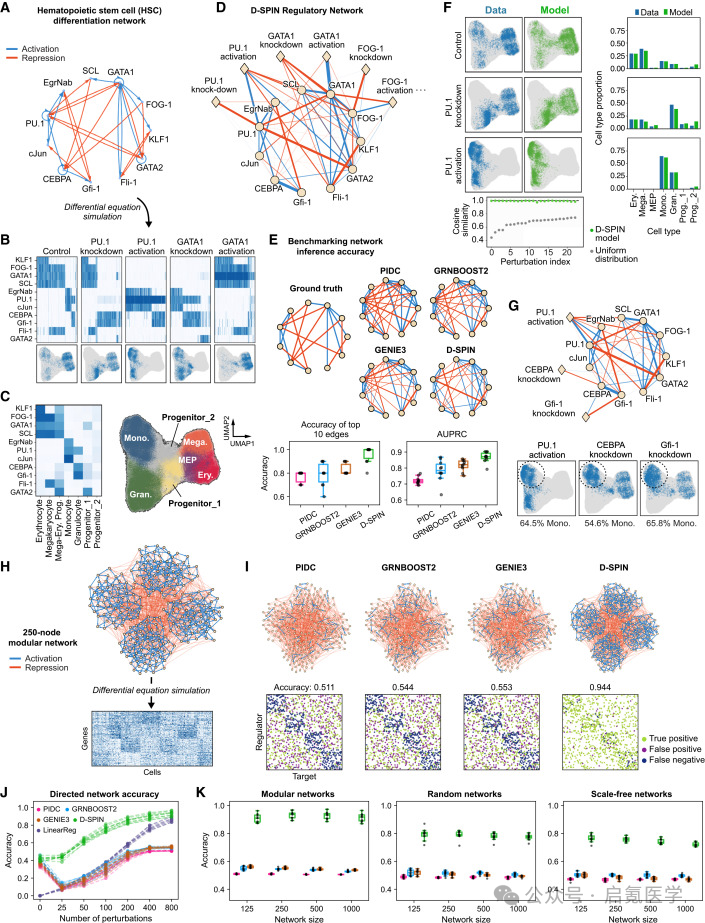
图2 D-SPIN在合成数据和大规模网络上的基准测试:(A)HSC分化网络的真实拓扑;(D-E)与PIDC/GRNBoost2/GENIE3的准确率对比;(H-K)从250到1000节点的大规模网络推断
502种药物×150万个细胞
团队还自建了一个大规模免疫调节药物数据集,是用抗CD3/CD28抗体激活人原PBMC(模拟T细胞驱动的免疫过度激活),然后加入502种小分子药物,收集了150万个过滤后的单细胞,覆盖1,200+个处理条件和28种免疫细胞状态。D-SPIN用465个网络交互参数 + 每个处理条件30个参数,就能高保真地模拟药物调控的细胞状态分布(超过90%样本的余弦相似度>0.9),并把70种有显著效应的药物自动分为7个表型类别,这与药物的已知生化靶点高度一致。
并且,基因级网络进一步揭示了不同药物类别的转录组特征,糖皮质激素选择性激活KLF9、TSC22D3、MAFB等GC受体信号和M2巨噬细胞极化相关基因;强抑制剂则更强地抑制STAT1/3、JAK3、IRF1/4/7/9等炎症通路基因。可以看出,D-SPIN是把笼统的“抑制”或“激活”拆解成了具体的基因调控回路。
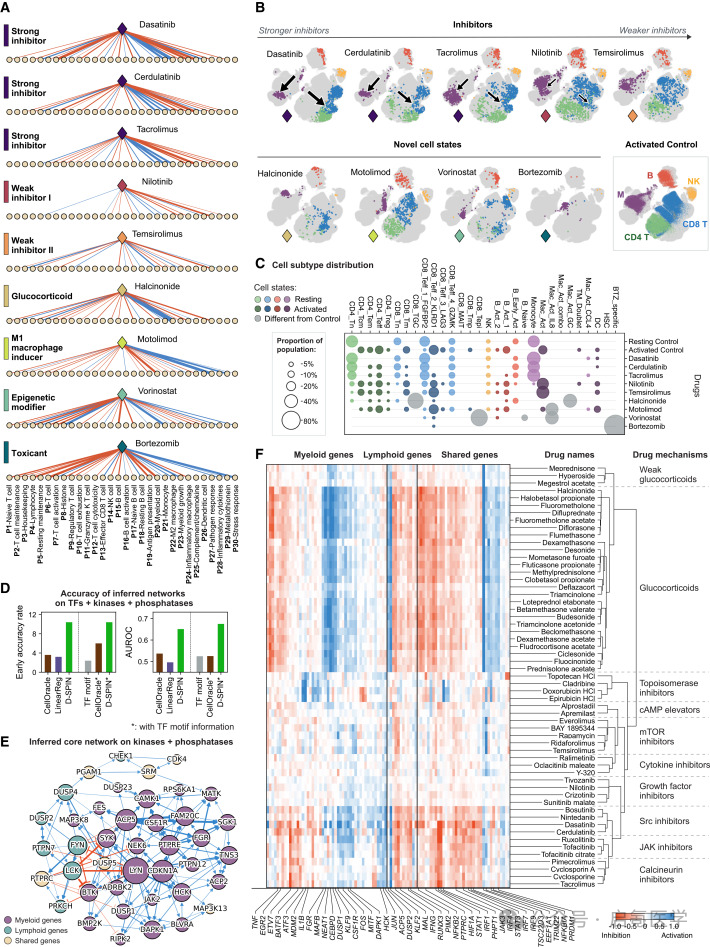
图3 D-SPIN将502种药物分为7个表型类别,并揭示不同药物机制的基因级特征:(A)药物分类树和UMAP可视化;(D)与CellOracle等方法的准确率对比;(F)不同药物类别的基因响应热图
1个组合实验
推断全局剂量响应
在药物组合分析中,团队选了10种不同类别的药物,实验测定了所有两两组合的响应。结果发现84%的药物交互作用在基因程序水平上是加性或次加性的,也就是说药物组合不是“1+1>2”的协同,而是“叠加招募”不同的基因程序来创造新的细胞状态。
以糖皮质激素(halcinonide)和Src抑制剂(dasatinib)的组合为例,两者都是抗炎药物,但通过完全不同的通路作用于M2巨噬细胞程序,也就是说它们激活同一个程序,但却用了两套独立的调控因子。这就说明了为什么组合能产生单药无法实现的“超抑制M2”巨噬细胞状态。
D-SPIN只需要单药剂量响应数据 + 一个组合条件,就能插值生成所有未观测剂量组合的细胞状态分布(从0个组合到加入仅仅1个组合条件,平均余弦相似度从0.72跳到0.84)。最终,D-SPIN绘制出了halcinonide和dasatinib剂量组合的髓系细胞状态“相图”。
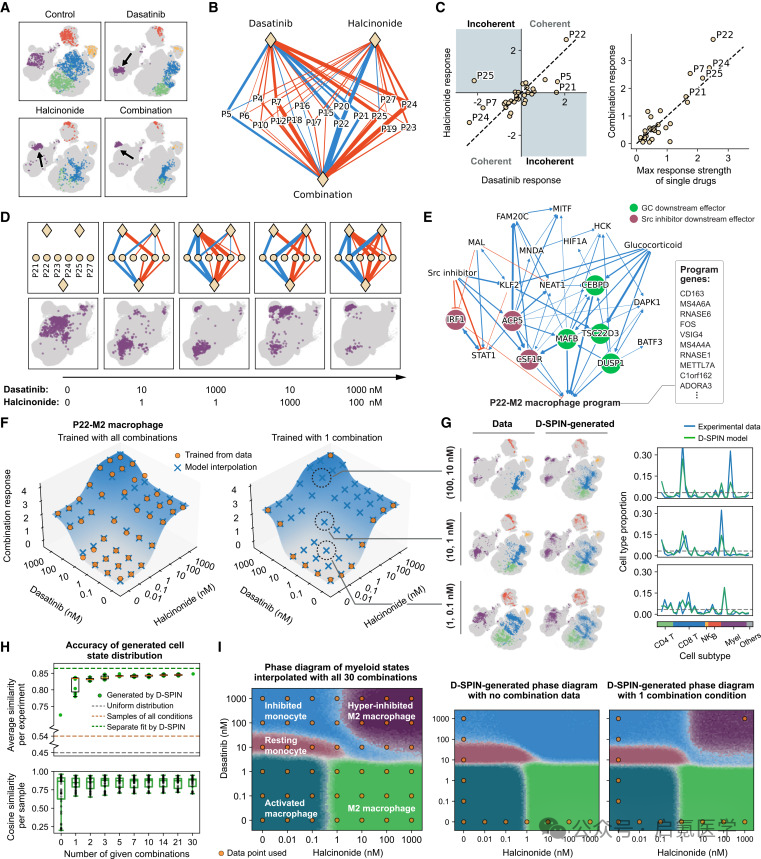
图4 药物组合的网络机制与剂量插值:(A-D)dasatinib/halcinonide组合剂量响应;(E)GC与Src抑制剂通过不同调控因子激活M2程序;(H)仅0到1个组合条件的插值精度跳跃;(I)髓系细胞状态相图
扰动分析的下一步
Ising这种统计物理模型的回归,使其平衡框架就像它曾经成功应用于神经网络和鸟群运动等非平衡系统一样,这次也很好的描述了细胞状态分。但显然细胞不是热平衡系统,所以研究团队也表示“为什么平衡模型能对这些显然远离热平衡的系统产生如此显著的预测力,这个问题仍然不清楚”。这也许暗示着细胞在某些层面上是可以被有效的建模为“被外力驱动通过不同配置的平衡系统”。
当然,目前D-SPIN也有其局限性,暂且只考虑到成对交互而没有高阶多体交互,属于平衡模型而无法处理动态过程,而且假设核心网络不会随扰动改变,因此不适用于涉及表观重编程的分化过程。不过,作为一个从扰动数据出发的可解释、可生成的基因调控网络推断框架,它在当下的Perturb-seq数据爆发期填补了一个关键的方法学空缺。如果你手边正好有需要做扰动分析的数据,不妨抓紧去试试。
DOI: 10.1016/j.cell.2026.04.028
GitHub: https://github.com/JialongJiang/DSPIN
文章改编转载自微信公众号:启氪医学
原文链接:https://mp.weixin.qq.com/s/srH9GDs8aMjlq9yA5BCIHw | 





















