本帖最后由 宇宙微尘 于 2025-11-12 02:02 编辑
蛋白–肽相互作用在信号转导、免疫调控和药物设计中具有核心作用。准确预测蛋白–肽复合物的结合构象与结合亲和力,是理解分子识别机制和推动肽类药物开发的关键。
2025年10月23日,华中科技大学黄胜友团队在Nat Mach Intell发表文章An interaction-derived graph learning framework for scoring protein–peptide complexes,提出了一种基于相互作用驱动的图学习框架——GraphPep,该框架能够从蛋白–肽复合物的三维结构中自动提取相互作用特征,并生成定量评分。GraphPep通过构建相互作用图并利用层次图神经网络建模复合物界面的几何与能量特征。在多个独立基准数据集上,GraphPep在结合能预测与构象评分任务中均显著优于现有方法,并展示出良好的可解释性与泛化性能。该框架可识别关键界面残基与能量热点区域,为基于结构的肽药设计提供新思路。

蛋白–肽相互作用(Protein–Peptide Interactions, PPIs)普遍存在于生物系统中,涉及酶调控、信号通路、免疫识别与基因表达控制等关键过程。肽分子因其高亲和力与可修饰性,正成为新一代药物设计的重要方向。然而,预测蛋白–肽复合物的三维结构与结合能仍充满挑战。传统的物理打分函数往往依赖经验参数,难以捕捉复杂的非线性界面特征。
随着深度学习的崛起,图神经网络(Graph Neural Networks, GNNs)已在蛋白–配体结合建模中显示出卓越能力,但肽分子高柔性、多构象、能量不均的特性,使得直接迁移变得困难。为解决这一问题,研究人员开发了GraphPep框架,以“相互作用”为核心表示单元,从几何、能量与拓扑层面联合建模蛋白–肽界面,从而提升模型物理一致性与预测精度。
方法
GraphPep包含三个关键模块:
相互作用图构建模块
基于蛋白–肽复合物结构生成相互作用图。节点表示原子或残基,边表示化学键及跨界面非键相互作用(如氢键、范德华接触)。通过距离与角度阈值定义蛋白与肽的跨界面连接关系,从而得到完整的“蛋白–肽相互作用图”。
特征编码与交互机制
每个节点和边包含原子类型、电荷、氢键供体/受体状态、疏水性与局部几何描述符。模型引入交互注意力机制(interaction-aware attention),以强化蛋白与肽间信息流动,使得模型能够捕捉到协同结构变化。
层次化图神经网络评分模块
使用多层图卷积网络(融合GIN与GAT)聚合节点特征并生成全局表示,输出复合物的结合评分。模型以实验结合能为监督信号,通过端到端学习优化参数,实现对复合物稳定性与亲和力的精准估计。
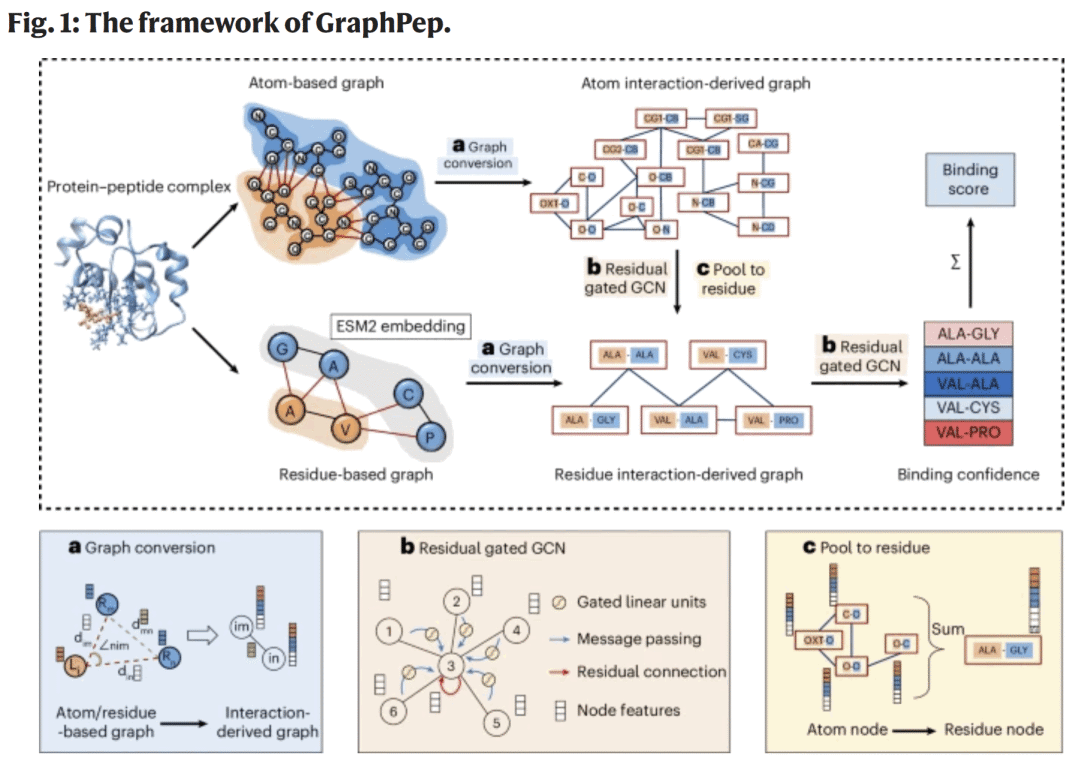
图1.GraphPep框架结构
结果
对接程序生成的诱饵集性能
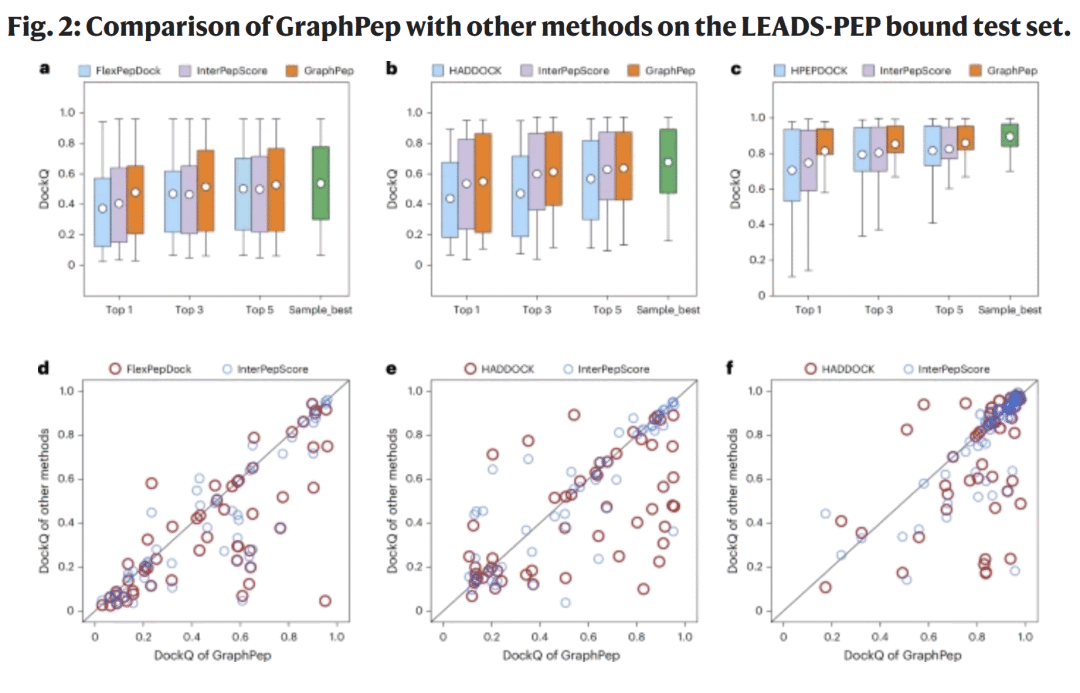
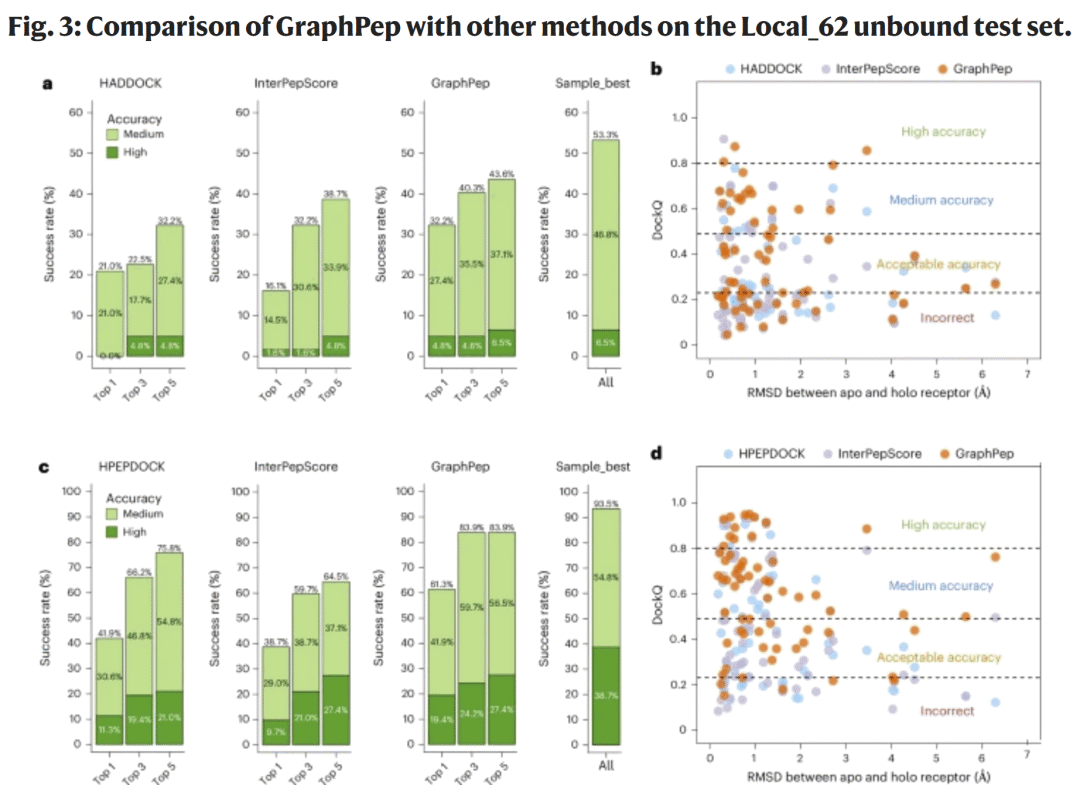
研究人员首先在LEADS-PEP(bound/unbound)与Local_62(unbound/unbound)基准集上,测试了GraphPep在FlexPepDock、HADDOCK、HPEPDOCK 生成的诱饵集上的表现。
结果显示,GraphPep在所有测试中均显著优于各对接程序自带打分函数与深度学习方法InterPepScore。例如,在HPEPDOCK诱饵集上,GraphPep在Top-1、Top-3、Top-5排名中的平均DockQ分数分别为0.814、0.854、0.860,均高于其他方法。进一步分析表明,GraphPep在多数复合物上能获得更高DockQ分数,并在无约束(unbound)结构条件下仍保持稳定性能。
在ADCP与AFM构建诱饵集上的表现
在ADCP99数据集上,研究人员利用ADCP与AlphaFold-Multimer(AFM)生成诱饵集,对GraphPep进行测试。在ADCP诱饵集中,GraphPep的中等精度与高精度成功率分别为56.6%与31.3%,显著高于ADCP(47.5%, 23.2%)与InterPepScore (51.5%,25.3%)。在AFM诱饵集中,GraphPep同样表现最优,并在共识评分策略下实现整体成功率74.7%,高于所有比较方法。这些结果表明GraphPep具有较强的泛化能力,能在不同诱饵生成机制下稳定识别真实构象。

AlphaFold3生成诱饵集上的表现
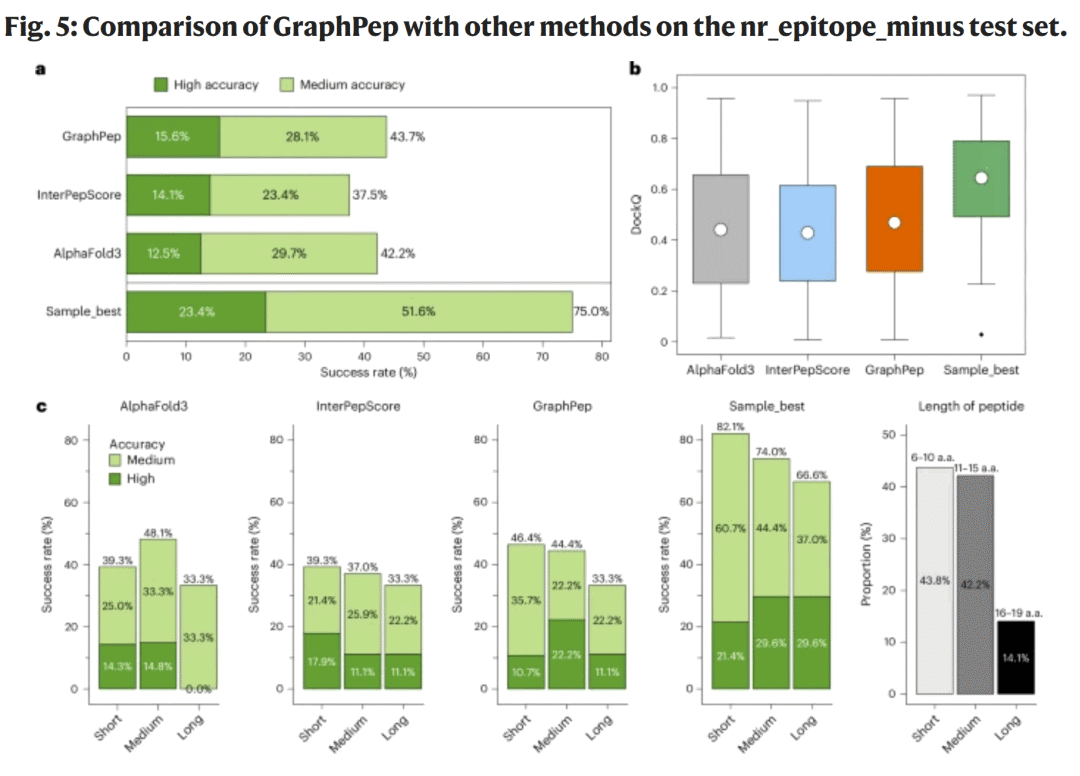
GraphPep进一步在nr_epitope_minus抗体–肽复合物集上验证性能。通过AlphaFold3强制采样生成的121组诱饵中,GraphPep在中等与高精度标准下的成功率分别为43.7%与15.6%,优于AlphaFold3原始评分与InterPepScore。结果显示,随着肽长度增加,预测准确率下降,但GraphPep对短肽与中等长度肽的表现始终领先。在DockQ平均分上,GraphPep为0.469,较AlphaFold3(0.441)与InterPepScore(0.428)均有提升。
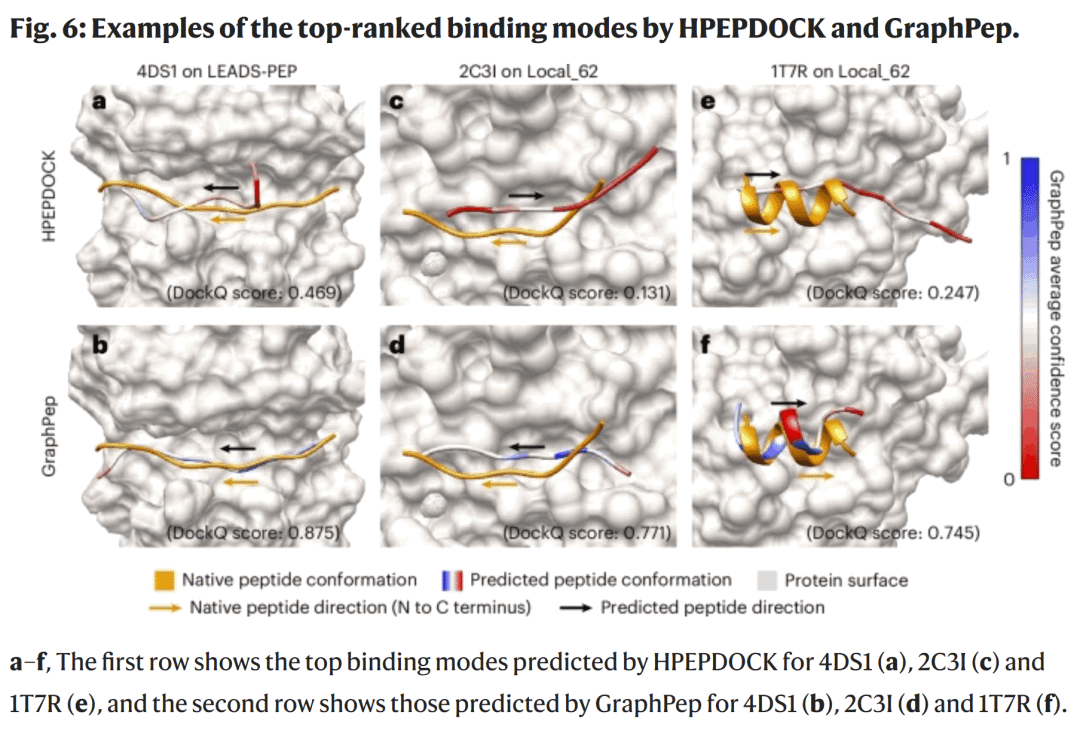
预测结合模式实例
研究人员展示了多个典型案例(如4DS1、2C3I、1T7R),对比HPEPDOCK与 GraphPep的预测结果。
GraphPep预测的结合构象与实验结构高度一致,能正确识别肽的方向、构象与二级结构类型。例如在4DS1中,GraphPep的DockQ为0.875,而HPEPDOCK仅为0.469。此外,GraphPep能为每对残基接触提供置信度评分,为后续结构优化提供指导依据。
讨论
GraphPep提供了一种以“相互作用”为核心的全新图学习思路,用于蛋白–肽复合物的构象评分与结合能预测。
其核心创新包括:
节点定义革新:以相互作用为节点,而非传统的原子或残基;
基于接触学习:以残基–残基接触为目标,减少对大量诱饵样本的依赖;
多尺度融合:结合原子层与残基层信息,平衡精度与鲁棒性;
语言模型嵌入:引入ESM-2表征进化信息,增强泛化能力;
物理一致性:模型学习出的能量曲线呈“漏斗型”分布,可用于构象优化。
GraphPep在多个独立数据集与不同来源诱饵上均表现优异,展现出广泛适用性。
此外,模型还能检测结构冲突、识别α-螺旋与非螺旋肽的不同结合特征,具备生物物理解释力。
研究人员认为,GraphPep可作为后续结构生成与肽药筛选的重要基础模型,并为未来结合生成式AI的蛋白–肽设计提供坚实框架。
参考资料
Tao, H., Wang, X. & Huang, SY. An interaction-derived graph learning framework for scoring protein–peptide complexes. Nat Mach Intell (2025).
https://doi.org/10.1038/s42256-025-01136-1
文章改编转载自微信公众号:智药邦
原文链接:https://mp.weixin.qq.com/s/4H5dpP1ao7B0T6nMQ81J3w | 





















