发表于 Cell 的《 A generative deep learning approach to de novo antibiotic design 》提出一种深度学习驱动的抗生素设计方法,以应对耐药菌感染的全球公共卫生威胁。该方法通过图神经网络 Chemprop 筛选 4 千万 + 化学片段,结合遗传算法 CReM 与变分自编码器 VAE,实现基于片段的扩展优化与从头分子设计。研究成功发现 NG1 和 DN1 两种新型化合物:NG1 对淋病奈瑟菌 MIC=0.5 μg/mL,靶向 LptA 蛋白;DN1 对金黄色葡萄球菌及多重耐药菌株具广谱活性,杀菌速度优于万古霉素。两者均拥有全新作用机制,在小鼠感染模型中验证有效,为耐药菌治疗提供了高效新路径,也推动了表型引导与生成模型结合的药物研发范式革新。
背景
当前,耐药性细菌感染已成为全球公共卫生的重大威胁。包括淋病奈瑟菌(N. gonorrhoeae)和金黄色葡萄球菌(S. aureus)在内的多种病原体,已产生对现有抗生素表现出广泛耐药性的菌株。由于耐药性细菌对现有抗生素的分子结构和作用机制具有广泛耐受性,因此研发具有新颖结构、全新作用机制的抗生素尤为关键。
传统抗生素研发在设计新结构、新机制药物分子上所需的成本和时间很高,从1980到2003年,头部的15家药企仅成功研发了5种新抗生素[1]。近年来,已有一些基于深度学习的方法用于抗生素发现,如基于图神经网络(Graph Neural Network, GNN)的分子性质预测模型,用于预测抗菌活性、细胞毒性等性质,以从虚拟化合物库中筛选分子。虚拟筛选受限于虚拟库大小,相比之下,生成式模型能够探索更大的化学空间,更具有发掘新结构分子的能力。
基于片段的药物发现(fragment-based drug discovery, FBDD)针对特定蛋白质靶标筛选大规模分子片段库,拓展了虚拟库的化学空间,是近年来药物设计的有力工具。相比之下,靶标无关(target-agnostic)的方法从表型(phenotype)测量出发,以整细胞生长抑制、毒性等可观察表型作为主要筛选指标,针对全细胞活性而非特定分子靶点进行筛选,从而整合不同化学型(chemotype)间有用的结构信息,产生具有多样作用机制的分子,并避免副作用,减少下游发现过程的损失率。基于表型的方法扩展了“可药”空间,可发现作用于传统靶标以外机制的分子,在发现新作用机制的药物上,相较于基于靶标的方法有显著的优势,尤其是在first-in-class药物发现过程中。目前基于表型的药物发现主要还是通过筛选方法进行,FAME(2022)是最早的基于表型的分子生成方法,它以药物诱导的基因表达数据训练模型自回归生成分子,但生成分子的实际有效性和可合成性并没有验证[2]。表型引导与基于片段方法的结合,以及和分子从头生成的结合,在抗生素发现领域都存在巨大探索空间。
近日,麻省理工学院(MIT)的James J. Collins等人发表在Cell上的文章《A generative deep learning approach to de novo antibiotic design》提出一种基于深度学习的抗生素化合物生成设计方法。该方法运用图神经网络(GNNs)基于表型筛选出活性分子片段,而后用遗传算法(CReM)和变分自编码器(VAE),从活性片段出发或从头生成抗菌化合物。作者对淋病奈瑟菌和金黄色葡萄球菌分别发现了NG1与DN1两种化合物,具有高活性和高选择性,且其作用机制与临床常用抗生素截然不同,并在小鼠感染模型中证实疗效。本研究构建的抗生素候选药物设计平台,不仅加速了抗菌化合物的发现进程,更为探索浩瀚未知的化学空间提供了高效路径。
方法与结果
2.1 抗淋病奈瑟菌(N. gonorrhoeae)的药物设计
2.1.1 优势片段筛选
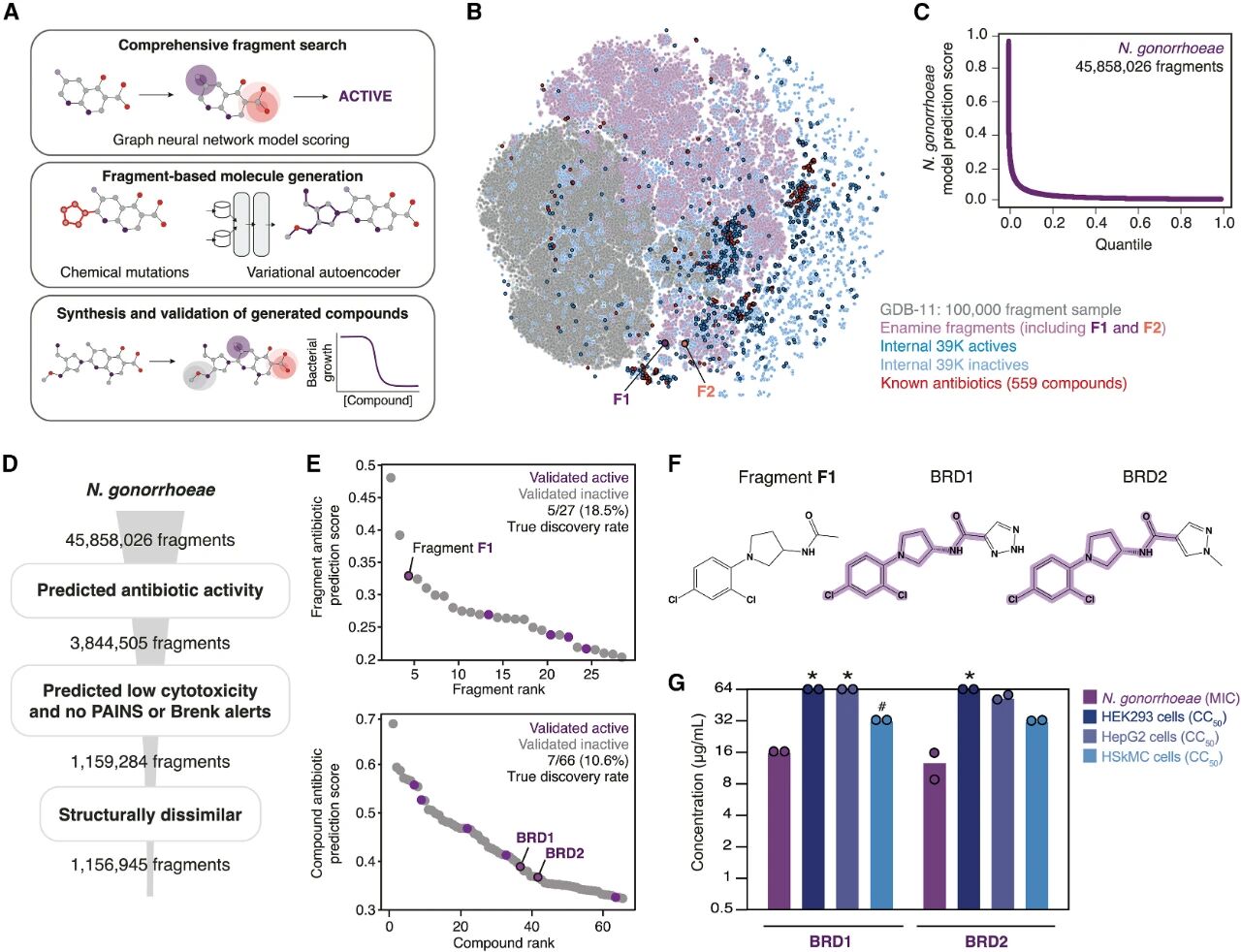
图1: 使用Chemprop筛选针对N. gonorrhoeae的优势片段。
在药物化学中,确定好优势片段是获得良好生物学活性的关键因素。基于这一经验,研究者首先利用基于图神经网络的Chemprop模型(见图1A),针对淋病奈瑟菌(N. gonorrhoeae)和金黄色葡萄球菌(S. aureus)进行了大规模片段筛选。作者整合了来自GDB-11、GDB-13与Enamine REAL 三大数据库的化学片段,构建了一个包含超过4千万个片段的虚拟筛选库。通过对上述化合物库结构分布的可视化(见图1B),作者发现其能够显著拓展已知抗生素的化学空间。
首先,针对N. gonorrhoeae,作者首先使用超过三万(38,765)条活性数据对Chemprop模型进行训练。利用该模型,作者筛选得到了约 384 万个具有潜在活性的片段(见图1C)。随后通过毒性预测、PAINS子结构筛选、与已知抗生素的相似性比对,作者获得了约115万个候选片段(见图1D)。
为验证筛选策略的有效性,作者在 79.9 万个已知活性的化合物中搜索包含候选片段的分子,发现了66 个化合物,其中7 个能有效抑制淋病奈瑟菌生长(真实发现率 10.6%,见图1E)。其中片段 F1 对应的两种活性化合物 BRD1 和 BRD2具有良好的抑菌能力,具有良好的发展潜力(见图1F-G)。
2.1.2 基于片段的分子生成
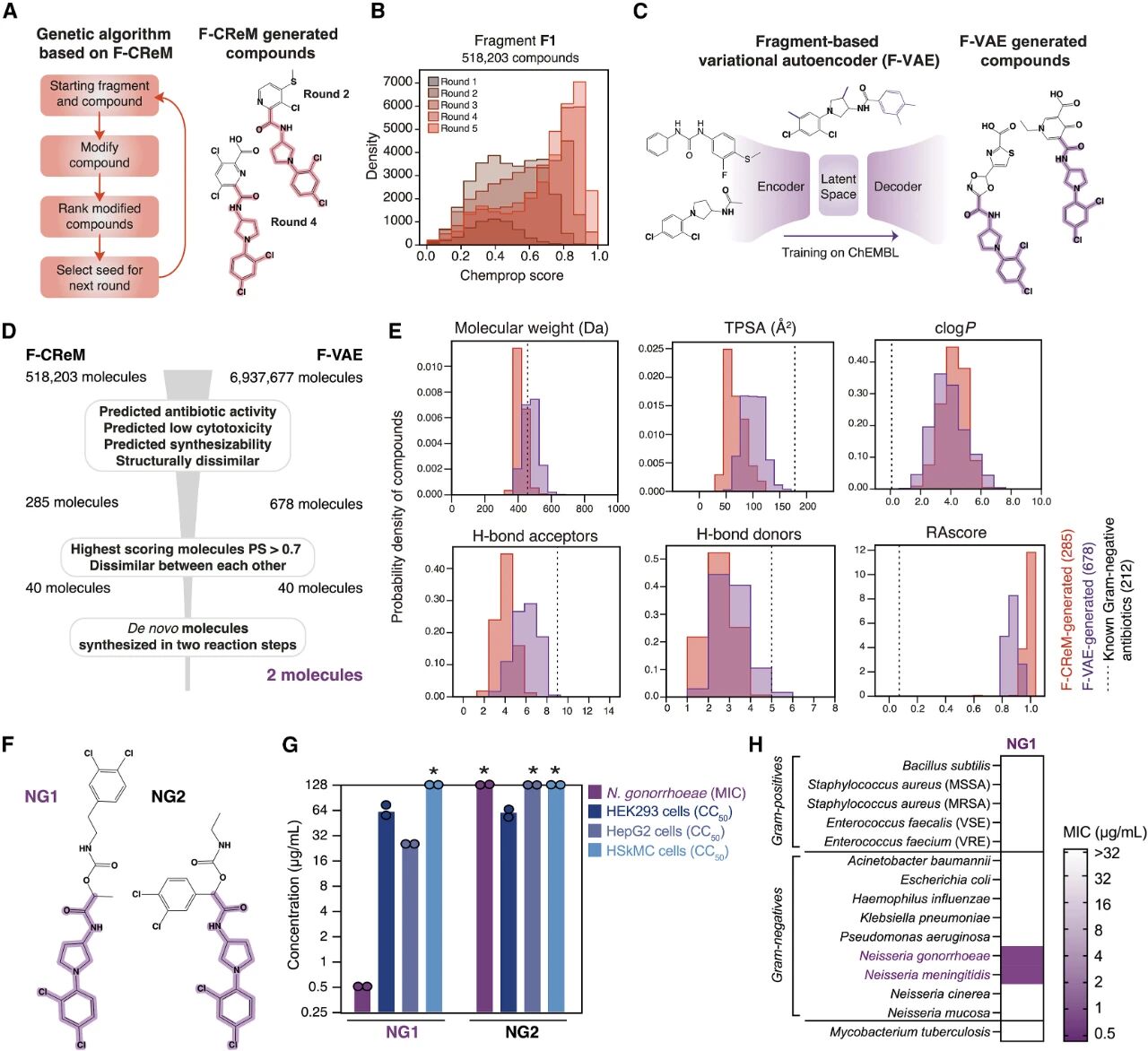
图2:使用生成模型从F1出发进行药物设计。
F1在可购买的化合物库中出现频率较低,因此作者决定使用分子生成模型基于F1进行设计。作者采用了两种生成式算法:基于遗传算法的 CReM(图2A)与片段驱动的变分自编码器 F-VAE(图2C)。对于CReM,作者将其与Chemprop结合,在每轮迭代过程中不断提升分子的预测活性(图2B)。F-VAE采用片段作为生长的起点,在生成过程中采用基于原子的生成方式。作者首先使用F-VAE基于F1片段进行大批量分子生成,而后使用Chemprop模型对生产结果进行筛选。
在完成基于CReM和F-VAE的分子生成后,作者对的到的分子进行了一系列后续筛选和聚类(图2D)。两种模型生成的分子在理化性质上有所差异,但在可合成性(RAscore)上相当(图2E)。最后,通过人工可合成性评估,作者完成了两个化合物,NG1和NG2,的合成(图2F)。其中仅 NG1 展现显著活性(MIC = 0.5 μg/mL,见图2G)。NG1的结构新颖、理化性质良好、且对耐药菌也有抑制活性(图3A),有较好的发展潜力。
2.1.3 NG1的作用机理分析

图3:NG1的机理研究以及体内实验结果。
通过分析NG1的抗菌谱(图2H),作者认为其可能通过新颖的机制发挥抗菌效应,并进行了机理研究。通过时间杀灭实验与最小杀菌浓度测定(图3B-C),作者发现NG1呈浓度依赖性杀菌作用,活性与阿奇霉素相当。通过对膜流动性的评价(图3D),作者发现 NG1 处理后细胞 Laurdan 荧光略有升高,提示其可能部分通过降低膜流动性发挥作用。进一步的 NPN 探针吸收试验(图3E)与 SYTOX Green 染色实验(图3F)显示 NG1 会显著影响细菌外膜通透性。
为确定NG1的潜在靶标,作者后续进行了PISA试验(proteome integral solubility alteration),发现NG1能够显著影响LptA(lipooligosaccharide (LOS) export system protein)的稳定性(图3H)。时间分辨的RAN测序发现NG1能够以剂量依赖的方式上调lptA的表达(图3H)。与多粘菌素 B 的协同实验(图3I)进一步验证了 NG1 对 LOS 生物合成通路的干扰。这些结果都提示 LptA 是其主要作用靶点。在小鼠实验中,NG1未表现出溶血性或致突变性,并在小鼠阴道感染模型中显示了良好的活性(图3J-K),这些结果展示了NG1的临床转化潜力。
为进一步提升NG1的活性并讨论构效关系,作者合成并测试了 74 个结构类似物,其中 8 个对淋病奈瑟菌具有活性。另有 4 个来自Enamine REAL Space 的类似物中,1 个表现出更高活性,且细胞毒性更低。其作用机制与 NG1 相同。图3L展示了作者总结的构效关系。
2.2 抗金黄色葡萄球菌(S. aureus)的药物设计
2.2.1 基于片段的筛选与设计
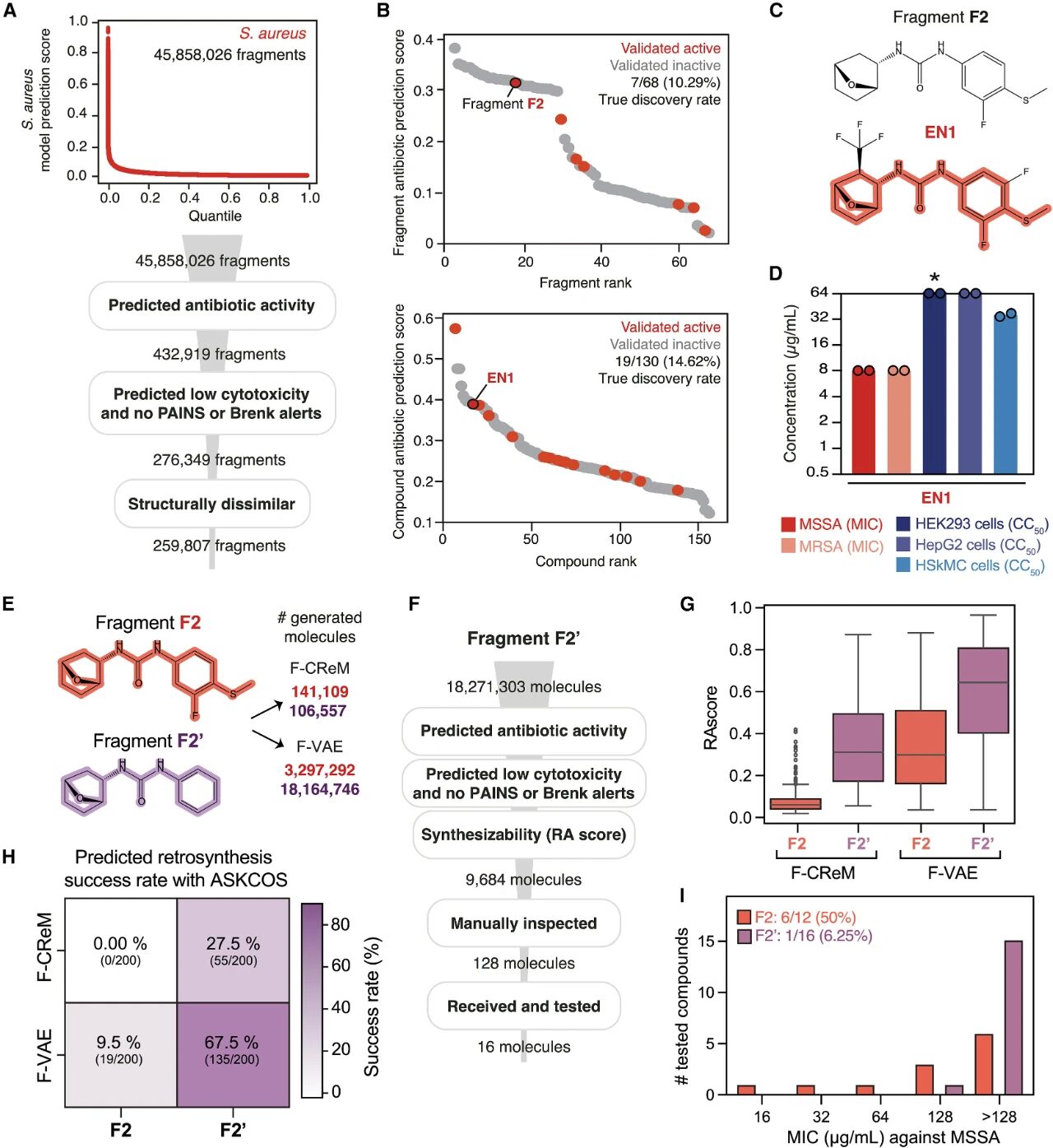
图4:使用基于片段的策略设计针对S. aureus的抗生素。
在成功设计针对N. gonorrhoeae的化合物后,作者将同样策略应用于针对S. aureus的抗生素设计中。作者首先采用了类似的策略,首先使用接近四万(39,312)条活性数据对Chemprop进行了训练,而后进行了分子骨架的虚拟筛选(图4A),并结合已有的测活结果筛选得到了骨架F2(图4B-C)。该骨架所对应的分子EN1对 MSSA 和 MRSA 的 MIC 为 8 μg/mL,且具良好选择性(图4D)。
F2片段在商业化合物库中的出现频率很低,因此作者决定采用基于CReM和F-VAE的分子生成范式来实现基于F2的药物设计(图4E)。不幸的是,直接基于F2设计的到的分子可合成性较差,因此作者将F2进行截断得到了F2‘(图4E),针对其设计的分子可合成性更高(图4G-H)。作者最终挑选了16个分子进行活性测试(图4F),其中仅一个具有活性,相比之下从Enamine REAL库中筛选得到的包含F2的12个分子中6个具有活性(图4I)。作者认为保持F2的完整结构对活性有重要的意义。
2.2.2 全新分子设计

图5:使用全新分子设计的方式开发针对S. aureus的抗生素。
为了获得抑制效果更好的分子,作者尝试摆脱初始片段的约束,采用全新分子设计的策略来获得高活性的分子(图5A)。作者对CReM和VAE的生成策略进行了修改,使得其能够实现从头设计(图5B-C)。评价结果显示基于JT-VAE的策略相比CReM能够生成物化性质良好的分子(图5D)。经多计算和人工的筛选(图5E),作者最终合成并测试 22 个分子,其中 6 个(DN1–DN6,图5G)表现出抗菌活性(命中率 27.3%,图5F)。其中,DN1具有最高的活性以及选择性(图6A)。
2.2.3 DN1-6的作用机理分析

图6:DN1-6的作用机理分析。
值得注意的是,DN1-DN6的结构存在较大的差异,抗菌机理可能不同(图6C-E),说明全新分子设计模型的确能够发现结构多样、作用机制不同的分子。作者对于活性较好的DN1进行了更细致的分析,包括膜流动性试验(图6F)以及形态学检测(图6G-H),发现DN1能够通过膜相关的机制来实现抗菌效果。
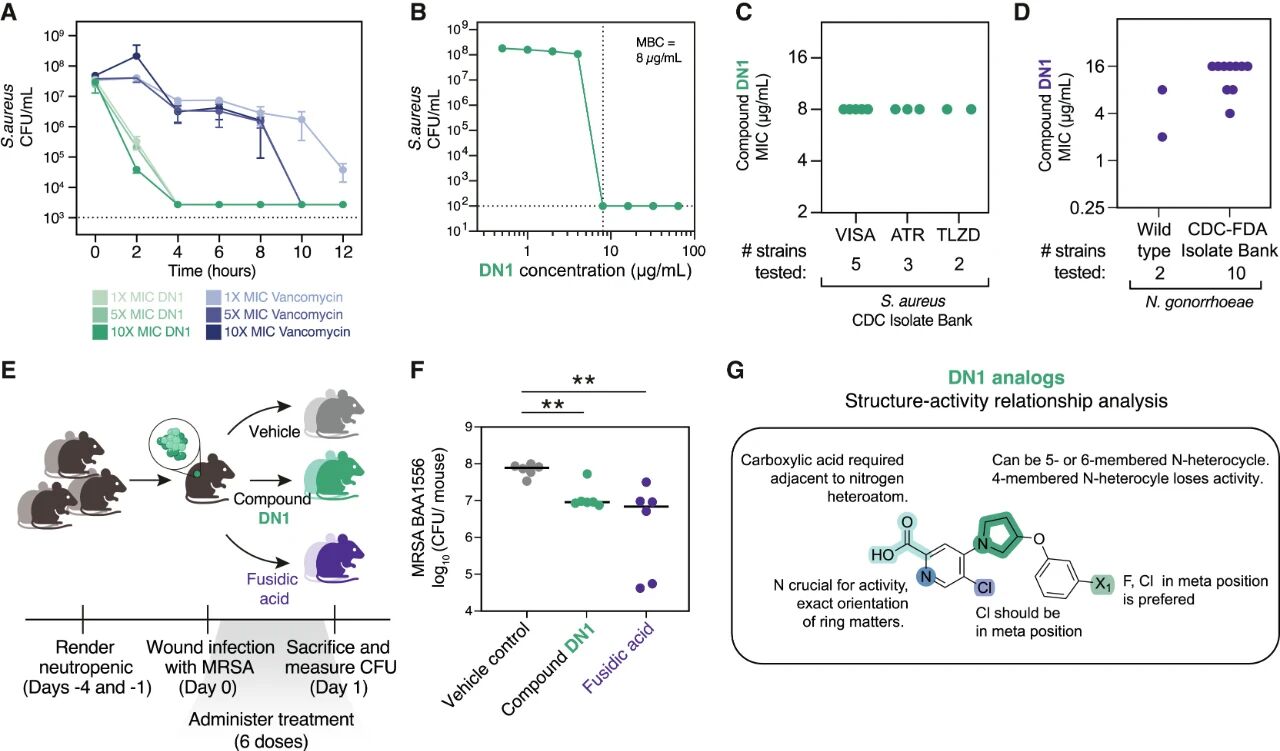
图7:DN1的体外、体内实验评价结果。
为评估 DN1 的临床转化潜力,作者进行了一系列的药效实验。时间杀灭实验显示,DN1 能在 2 小时高效杀灭S. aureus,速度快于临床常用的万古霉素(图7A-B)。DN1 还对 CDC-FDA 抗药菌株库中 10 株多重耐药革兰氏阳性菌(包括 VISA、ATR、TLZD 型菌株)均具抑制活性(图7C),且能抑制耐药性N. gonorrhoeae(图7D),显示其良好的广谱抗菌效能。在小鼠的皮肤感染模型上(图7E),作者发现DN1有与夫西地酸相当的抑菌能力(图7F),展示了DN1良好的临床应用前景。最后,针对DN1进行了衍生物合成与评价,对构效关系进行了分析(图7G),并发现了活性更优的衍生物DN1–164。
小结
本研究通过深度学习驱动的化合物生成设计,成功发现了针对高耐药性的淋病奈瑟菌和金黄色葡萄球菌的新型抗菌分子。研究首先利用图神经网络(GNN)作为评分函数,对超过4500万个化学片段进行虚拟筛选,识别出具有选择性抗菌潜力的起始片段。在此基础上,采用基于遗传算法的CReM模型和变分自编码器(VAE)模型进行分子生成,既支持基于片段的扩展优化,也能实现从头分子设计。在最终合成验证的24种化合物中,两种代表性化合物NG1和DN1表现出显著抗菌活性:NG1对致病性奈瑟菌属呈现窄谱活性,而DN1对革兰氏阳性菌及淋球菌均具有广谱抑制效果。重要的是,这两种化合物均展现出与传统抗生素不同的作用机制,在多个感染动物模型中有效降低细菌负荷,并具备清晰的构效关系景观(structure–activity landscape),为后续结构优化奠定了基础。
本研究采用以片段为起点的生成,能够全面探索分子构建单元,在此基础上连接组合,探索更大化学空间,并保持分子的稳定性、类药性;基于表型进行片段筛选,跳过靶标以细菌生长抑制为指标,有望让分子规避现有抗菌耐药机制,发现新作用机制。在两种分子生成模型中,CReM 依赖于基于规则的生物活性分子特征,通过“结构生长”或“结构修饰”来生成结构多样的分子,而 VAE 模型在生成结果上更具药类药性(drug-like)与可合成性(synthesizable)。本研究构建的模块化平台易于集成其他生成模型,如生成对抗网络(GAN)、流模型(Flow-based models)、扩散模型(Diffusion models)等,实现策略互补。
本研究开发的方法仍存在假阳性结果、合成可行性的问题,需要结合更精准的可合成性预测工具和逆合成算法,以及活性验证实验。未来该研究还将引入多目标优化框架,同时对抗菌活性、可合成性、低细胞毒性、代谢稳定性等进行优化,推动结构更新颖、具有新作用机制的抗生素发现。
参考文献:
原文:Krishnan, Aarti et al. "A generative deep learning approach to de novo antibiotic design." Cell, 188(21):5962-5979.e22(2025). https://www.cell.com/cell/fulltext/S0092-8674(25)00855-4
[1] Vincent F., et al. "Phenotypic drug discovery: recent successes, lessons learned and new directions." Nat Rev Drug Discov. 21(12):899-914 (2022). doi: 10.1038/s41573-022-00472-w.
[2] Pham TH., et al. "FAME: Fragment-based Conditional Molecular Generation for Phenotypic Drug Discovery." Proc SIAM Int Conf Data Min. 2022:720-728 (2022). doi: 10.1137/1.9781611977172.81.
文章改编转载自微信公众号:GoDesign
原文链接:https://mp.weixin.qq.com/s/VBwXDQR0ieKapVP3qSKQfw?scene=1
作者:李亦博 凌心辽
审稿:黄志贤
编辑:黄志贤 | 





















