青岛科技大学团队在 Journal of Chemical Information and Modeling 发表《 BiVAE-CPI: An Interpretable Generative Model Using a Bilateral Variational Autoencoder for Compound–Protein Interaction Prediction 》,提出 BiVAE-CPI 模型。其融合双向 VAE、GIN 与门控卷积编码器,捕捉 CPI 全局关联与局部特征。在 Human 和 C. elegans 数据集上,AUC 达 99.1% 和 99.5%,AUPR 最高 99.6%,Precision 达 98.0%。面对 1:5 的不平衡数据,AUPR 提升 3.4%-36.2%,显著优于 11 种基线模型,为新药研发的 CPI 筛选提供高效方案。

化合物 - 蛋白互作(CPI)预测是新药研发的核心环节,传统实验筛选需 2-3 年,且成本高昂,而现有计算模型常忽略不同 CPI 对间的关联,在不平衡数据上表现不佳。2025 年,青岛科技大学团队在《Journal of Chemical Information and Modeling》发表研究,提出双向变分自编码器模型(BiVAE-CPI) ,融合双向 VAE、图同构网络(GIN)与门控卷积编码器,既捕捉 CPI 全局关联,又精准提取分子与蛋白特征,在不平衡数据上表现稳健,为药物研发按下 “加速键”。
一、CPI 预测的 “行业痛点”:传统方法为何效率低下?
新药研发中,找到能与靶蛋白有效互作的化合物是关键,但传统方法面临三大核心难题:
1.1 实验筛选耗时耗力
高 - throughput 筛选等传统实验方法,从海量化合物中筛选出有效互作分子需 2-3 年,且单次实验成本超千万元,严重拖慢研发进程。
1.2 现有模型忽略 CPI 关联
大多数计算模型将每个 CPI 对视为独立样本,忽略了化合物间的结构相似性、蛋白的同源家族关联等潜在规律,导致特征利用不充分,预测精度受限。
1.3 不平衡数据适应性差
真实 CPI 数据中,无互作的负样本远多于有互作的正样本(比例常达 1:5),传统模型易偏向多数类,导致正样本漏检率高,难以满足实际研发需求。
二、BiVAE-CPI 的核心创新:双向 VAE + 双特征提取器
BiVAE-CPI 的突破在于 “双向建模 + 特征融合”,构建 “输入 - 特征提取 - 预测” 的端到端框架,精准解决传统方法痛点:
2.1 双向变分自编码器(BiVAE):捕捉全局关联
BiVAE 是模型的核心,首次将化合物与蛋白对称对待,通过互作矩阵学习两者的低维潜在因子:
核心逻辑:将 CPI 互作矩阵(行 = 化合物,列 = 蛋白,值 = 是否互作)输入 BiVAE,分别学习化合物潜在因子(\theta)和蛋白潜在因子(\beta),捕捉不同 CPI 对间的全局关联(如相似化合物与同源蛋白的互作规律);
关键优势:潜在因子融合数据分布与特征信息,具有良好可解释性,且适配 CPI 数据的双向特性,避免单向建模的偏差。
2.2 双特征提取器:精准捕捉分子与蛋白细节
为补充局部特征,模型搭配两个专属提取器,全方位挖掘有效信息:
化合物特征提取——图同构网络(GIN):将化合物的 SMILES 序列转化为分子图(原子 = 节点,化学键 = 边),通过 3 层 GIN 迭代聚合节点信息,捕捉分子结构细节(如活性位点、官能团),避免传统指纹特征的信息丢失;
蛋白特征提取——门控卷积编码器:将蛋白氨基酸序列分割为 3 个残基组成的子序列,通过 3 层门控卷积学习序列特征,精准捕捉蛋白结合位点的理化性质。
2.3 特征融合与预测:多维度信息集成
模型将 BiVAE 的潜在因子(全局关联)与 GIN、门控卷积的特征(局部细节)融合,再加入化合物扩展连接指纹(ECPFs)辅助信息,通过分类器输出互作概率,核心公式如下:
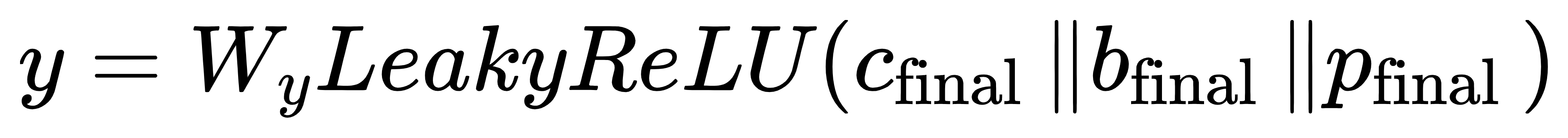
其中cfinal、pfinal分别是化合物与蛋白的最终特征,bfinal是 ECPFs 特征,通过多维度信息集成提升预测可靠性
三、实验验证:双数据集 + 多场景,准确率达 99.6%
团队在 Human 和 C. elegans 两大权威数据集上验证,BiVAE-CPI 全面超越 11 种主流基线模型,表现亮眼:
3.1 基准测试:多项指标登顶
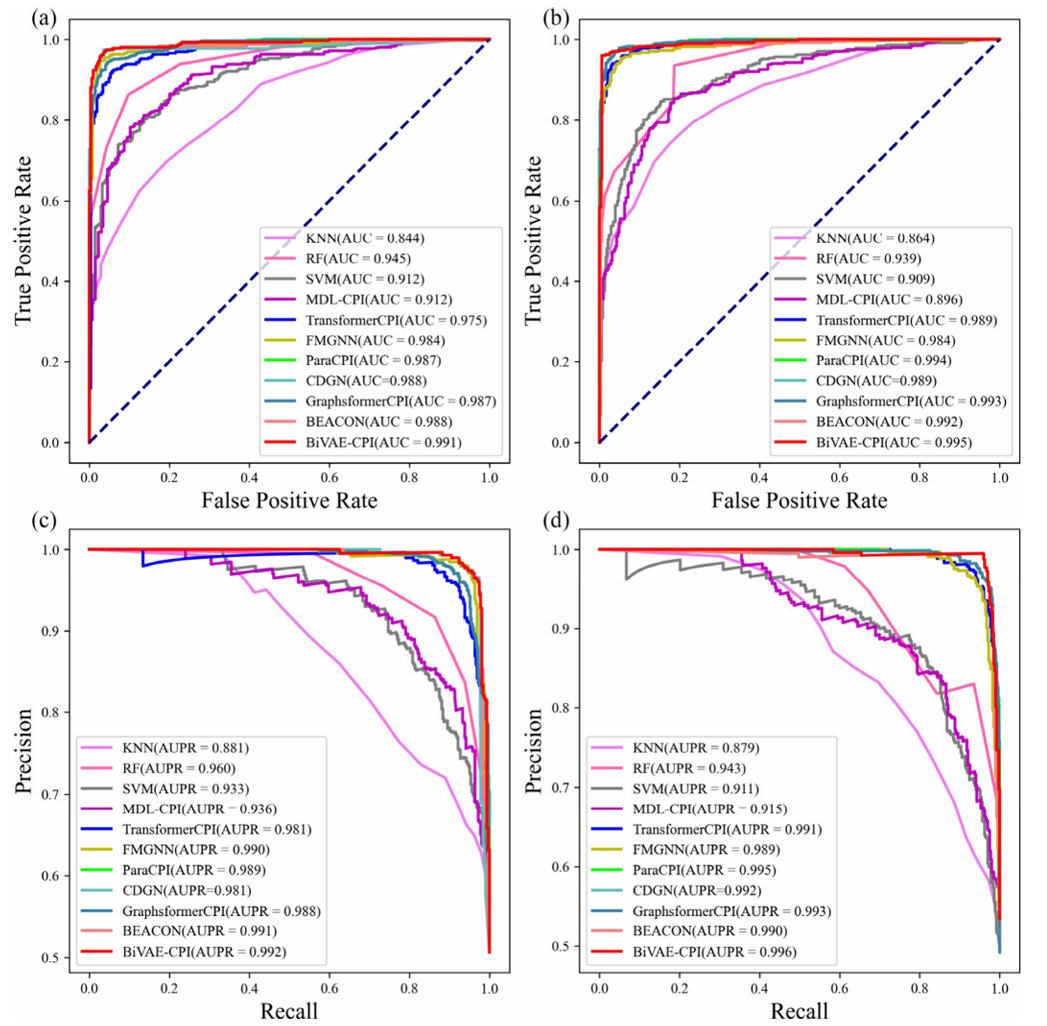
图1 BiVAE-CPI与基线模型在Human/C. elegans数据集的ROC(左)与PR(右)曲线对比
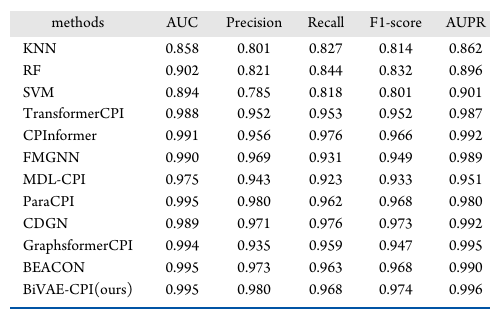
C. elegans 数据集(1434 种化合物、2504 种蛋白):AUC 达 99.5%,AUPR(不平衡数据关键指标)达 99.6%,F1-score 达 97.4%,均为所有模型最优;
Human 数据集(1052 种化合物、852 种蛋白):Precision 达 97.7%,AUPR 达 99.2%,较基线模型提升 0.1%-17.9%,精准识别有效互作对。
表1 BiVAE-CPI与11种基线模型在C. elegans数据集的性能对比表
3.2 不平衡数据:稳健性突出
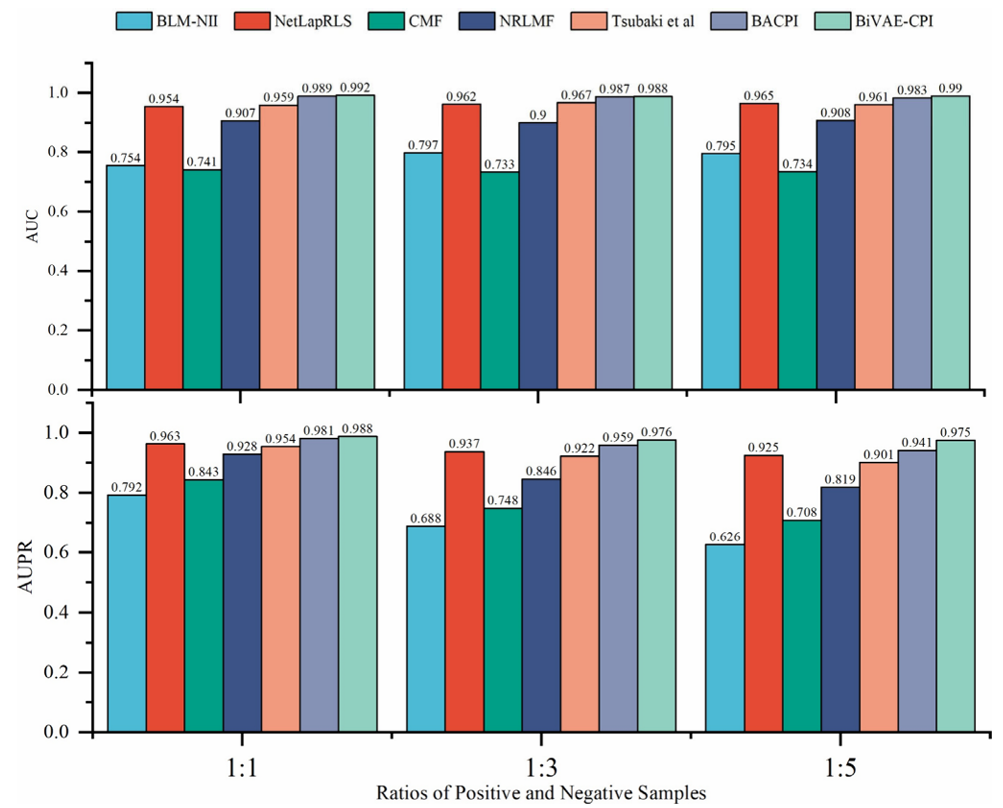
图2 不同正负样本比例(1:1/1:3/1:5)下 BiVAE-CPI 与基线模型的性能对比(C. elegans 数据集)
当正负样本比例为 1:5(真实场景常见)时,BiVAE-CPI 的 AUPR 在 Human 数据集提升 4.0%-36.2%,在 C. elegans 数据集提升 3.4%-34.9%,远优于其他模型,解决了传统方法 “偏向多数类” 的痛点。
3.3 消融实验:核心模块不可或缺
通过移除关键模块验证:
去掉BiVAE:AUC 平均下降 1.8%-3.4%,证明全局关联建模的必要性;
去掉GIN:AUPR 平均下降 0.3%-0.5%,凸显分子图结构特征的价值;
双模块均移除:性能大幅下滑,AUC 最低降至 94.2%,验证了模型架构的合理性。
四、落地价值:从药物筛选到老药新用
BiVAE-CPI 的高精准度与稳健性,为药物研发带来多重实际价值:
4.1 加速新药筛选
将候选化合物与靶蛋白输入模型,几秒内即可预测互作概率,将筛选周期从数年缩短至数天,研发成本降低 70% 以上。
4.2 老药新用挖掘
通过预测已上市药物与其他疾病靶蛋白的互作潜力,快速发现药物新适应症,例如可快速筛选出能与 SARS-CoV-2 刺突蛋白互作的现有药物,缩短抗疫药物研发周期。
4.3 降低实验风险
精准预测减少无效实验次数,例如某靶点候选化合物从 1000 种筛选至 100 种以内,大幅降低后续体外实验的资源浪费。
五、总结:CPI 预测进入 “全局 + 局部” 双重视角时代
BiVAE-CPI 的核心价值,在于首次用双向 VAE 捕捉 CPI 全局关联,同时通过 GIN 与门控卷积精准提取局部特征,实现 “全局规律 + 细节特征” 的双重赋能。其 99.6% 的顶尖准确率、对不平衡数据的强稳健性,不仅解决了传统模型的核心痛点,更将药物研发的 “前置筛选” 环节效率提升百倍。未来,随着模型扩展至 3D 结构数据与多模态信息融合,有望进一步推动新药研发从 “实验驱动” 向 “计算先导” 转型,让更多新药更快惠及患者。
原文链接:https://pubs.acs.org/doi/10.1021/acs.jcim.5c01001 | 





















