|
2026年02月25日,华东理工大学药学院/华东师范大学药学院、人工智能新药创智中心李洪林团队、华东师范大学计算机学院张凯团队,联合复旦大学类脑智能科学与技术研究院张捷团队等多家单位在Nature Machine Intelligence发表题为“Conditional diffusion with locality-aware modal alignment for generating diverse protein conformational ensembles”的研究论文。

该研究提出了一种名为Mac-Diff的条件扩散模型,用于生成蛋白质的构象分布,加速MD采样,并能有效预测蛋白质的构象异质性。该模型通过创新设计的局部感知模态对齐注意力模块,有效融合了物理先验、残基对空间关系及进化驱动的序列表征。在生成蛋白质构象分布方面,Mac-Diff不仅在采样效率上显著优于传统分子动力学 (MD) 模拟,更能有效探索蛋白质构象空间,在准确性与多样性的平衡等多个指标展现出色性能。在探索蛋白构象异质性方面,Mac-Diff在多个测试蛋白质案例上成功捕获了功能相关的构象态,综合表现优于绝大多数现有方法。这项研究体现了人工智能与计算生物学的深度融合,展示了生成模型在加速分子模拟方面的巨大潜力,不仅有助于揭示蛋白质功能相关的复杂动态行为,也为靶向瞬时构象的药物设计提供了新的高效工具。
研究背景
蛋白质作为生命活动的基本执行单元,其内禀灵活性是发生构象转换,进而行使生物学功能的基础。然而,捕捉这种动态特性一直面临巨大挑战:(1) 传统实验方法通常只能解析单一静态结构,难以捕捉瞬态构象及其变化过程。(2) 尽管MD模拟被广泛用于采样蛋白质连续的动态轨迹,但受限于积分步长和采样时间尺度等原因,计算过程仍昂贵耗时。(3) 近年来,以AlphaFold2为代表的模型通过端到端的整合结构和共进化信息显著提升了蛋白质结构预测的精度,但其输出仍偏向于热力学上最稳定的结构。最近,一系列基于AlphaFold2的变体通过扩展AlphaFold2的输入(MSA、模板等)及输出以探索蛋白质的功能构象。但其通用性及表现仍有待进一步验证和提升。另一方面,以扩散方法为代表的生成模型在构建蛋白质的几何表示、设计去噪网络及引入序列表征作为条件信息时,大多依赖于结构预测模型的组件。考虑到结构预测模型的优化目标是PDB数据库中单一的稳态结构,这种依赖性可能会引入归纳偏置,进而限制生成模型对多个亚稳态构象空间的探索能力,导致预测结果偏向于单个主导结构。基于上述背景和研究现状,能否从序列表征中提取足够的信息以解决构象异质性问题仍有待进一步探索,此外,目前针对上述挑战已开发的模型仍显不足。因此,如何设计一种既能利用大规模预训练模型的无偏序列信息,又能有效探索蛋白质构象景观的生成模型,是当前领域仍需解决的关键科学问题之一。
研究内容
研究团队开发了一种基于分数的“模态对齐条件扩散算法”——Modal-aligned conditional Diffusion (Mac-Diff) ,用于快速生成真实且多样的蛋白质构象集合(图1)。Mac-Diff算法的设计包括三个主要组成部分:1) 局部感知的模态对齐注意力 (Locality-Aware Modal Alignment-attention, LAMA-attention) 模块基于物理先验,将蛋白质语言模型ESM-2的序列表征和残基对的空间几何信息进行深度对齐,使模型能够敏锐感知蛋白质局部环境的变化。2) 适配的三角乘法更新机制,进一步在特征空间内显式建模残基间的几何依赖关系,强化残基对表征的几何关系建模。3) 多尺度去噪网络:采用UNet架构作为骨干网络,将上述更新后的残基对表征整合进蛋白质几何表示中,通过多轮逆向过程逐步去除噪声,最终还原出蛋白质真实的结构表示。

图1 Mac-Diff模型架构。(a) 蛋白质的几何表示。(b) UNet去噪网络。(c) 局部感知的模态对齐注意力模块[1]
为直观阐明LAMA-Attention的设计精髓,作者对比了其与经典的文生图模型的差异(图2)。传统的交叉注意力机制在像素和词语间建立全局密集的链接,缺乏明确的结构化对应(图2a)。相比之下,Mac-Diff紧密围绕蛋白质特有的生物学属性,对蛋白质不同模态之间的信息交互机制进行了更加深度的适配,通过物理先验实现了注意力的“去冗存精”。模型通过将注意力场精准聚焦于残基的局部相互作用邻域,仅聚合生物学上存在交互的残基特征。这种位置感知的空间对齐策略,确保了序列信息向结构空间的高效注入,显著提升了模型对蛋白质局部动态的捕捉能力。

图2 交叉注意力机制和LAMA注意力机制示意图[1]
作者将Mac-Diff在由12个快速折叠蛋白MD轨迹构成的测试集上进行综合评估。结果表明,与6个先进方法相比,Mac-Diff在JS-PwD、JS-Rg、JS-TIC等多个衡量分布差异的关键指标上展现出色的性能(图3a)。同时,Mac-Diff有效兼顾了生成样本的多样性-保真度性能(图3b)。在2D-TIC空间及残基对接触概率的表现进一步表明Mac-Diff能够生成高度逼真的构象分布(图4)。
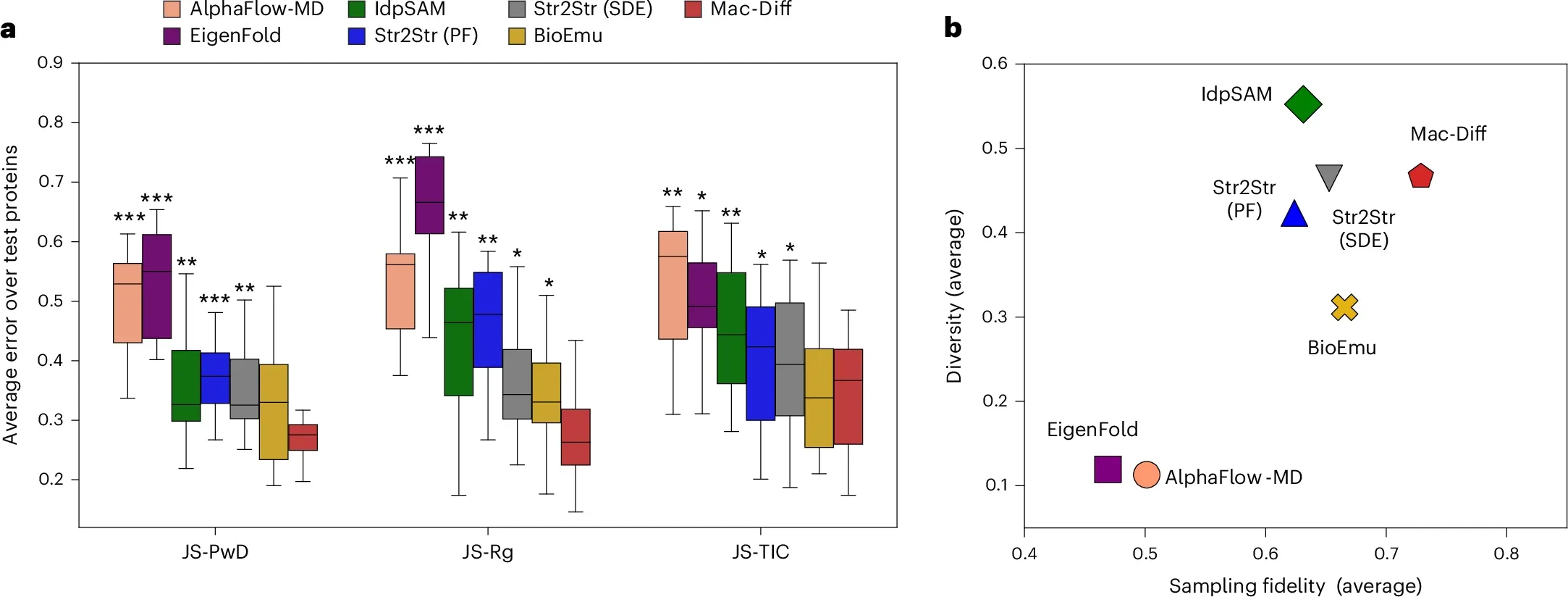
图3 Mac-Diff的采样性能。(a) 模型生成构象分布和真实MD分布的JS散度。(b) 采样保真度-多样性性能[1]

图4 Mac-Diff预测性能。(a) 模型采样构象在2D-TIC空间投影。(b) 残基接触概率分析[1]
此外,针对经典的蛋白质BPTI,作者测试了Mac-Diff模型和现有方法在探索蛋白质构象空间的能力。Mac-Diff生成构象不仅覆盖范围更广(图5),且在5个构象簇上的平均RMSD值最优。特别是在构象簇4、5以及远端的构象簇3上,Mac-Diff均取得了最低的RMSD,更精准恢复了罕见构象。
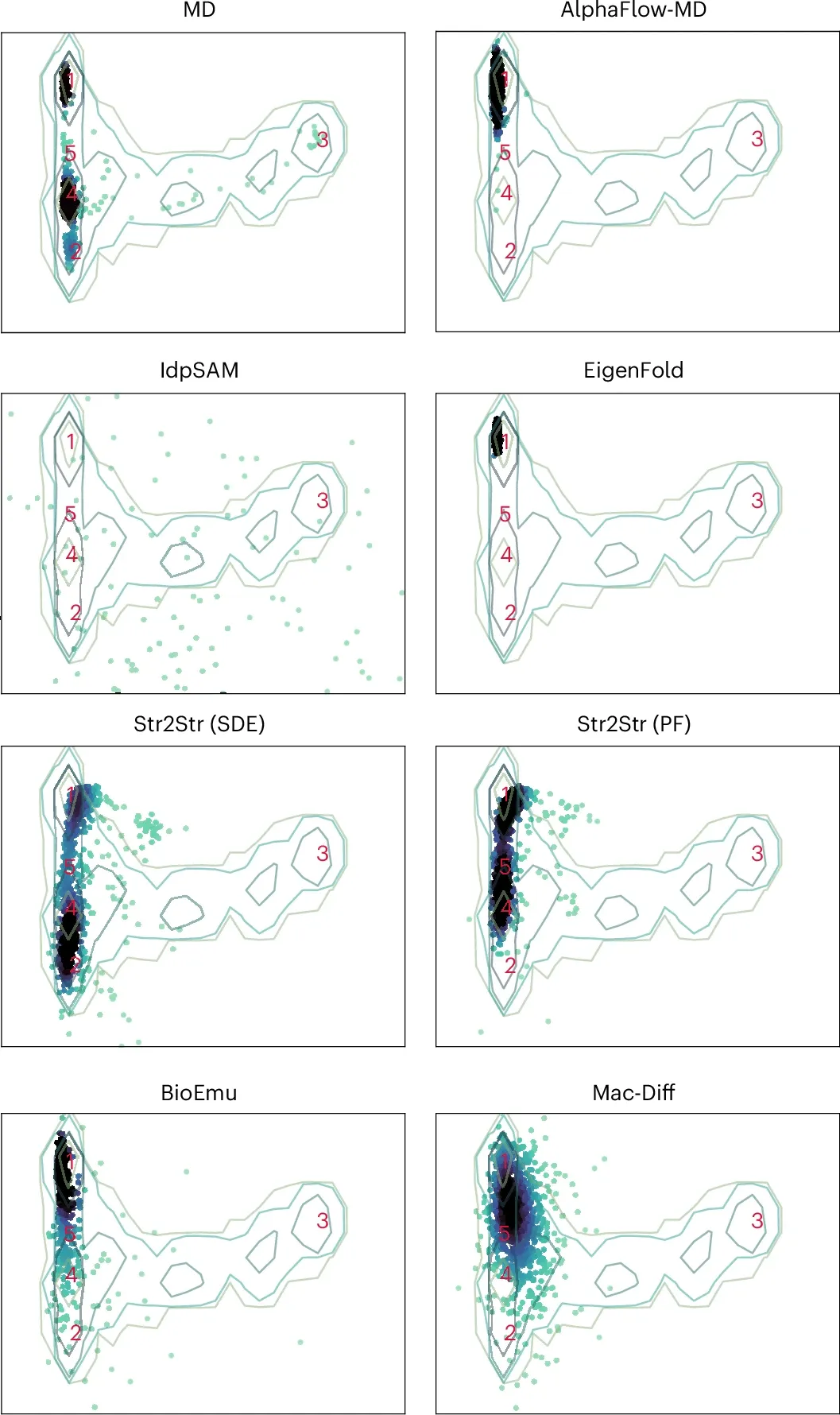
图5 Mac-Diff生成构象集合在2D-TIC空间的投影。MD轨迹中的5个代表构象以数字表示[1]
为了进一步验证模型在真实生物学场景下的价值,作者在腺苷激酶AdK案例和Cfold40测试集上深入探究了模型对蛋白质功能相关构象态的捕获能力。对比8种竞争方法:(1) 在变构蛋白AdK案例中,Mac-Diff不仅成功采样到低能的闭合构象,而且敏锐的捕获了开放构象(图6a);(2) 在TAF1的串联溴结构域、蛋白质二硫键异构酶的b’–a’域、跨膜蛋白OpuA的底物结合域、串联VHS和FYVE结构域和表面层蛋白5个代表案例上,Mac-Diff均能有效探索两个功能构象态(图6b-f)。(3) 在综合排名中,Mac-Diff在平均TM-score 和RMSD指标上的综合表现优于绝大多数竞争方法(图6g)。这表明 Mac-Diff 通过引入有效的局部感知的模态对齐注意力机制,展现出色的构象生成能力,为未来探索蛋白质动力学提供了关键的技术支撑。
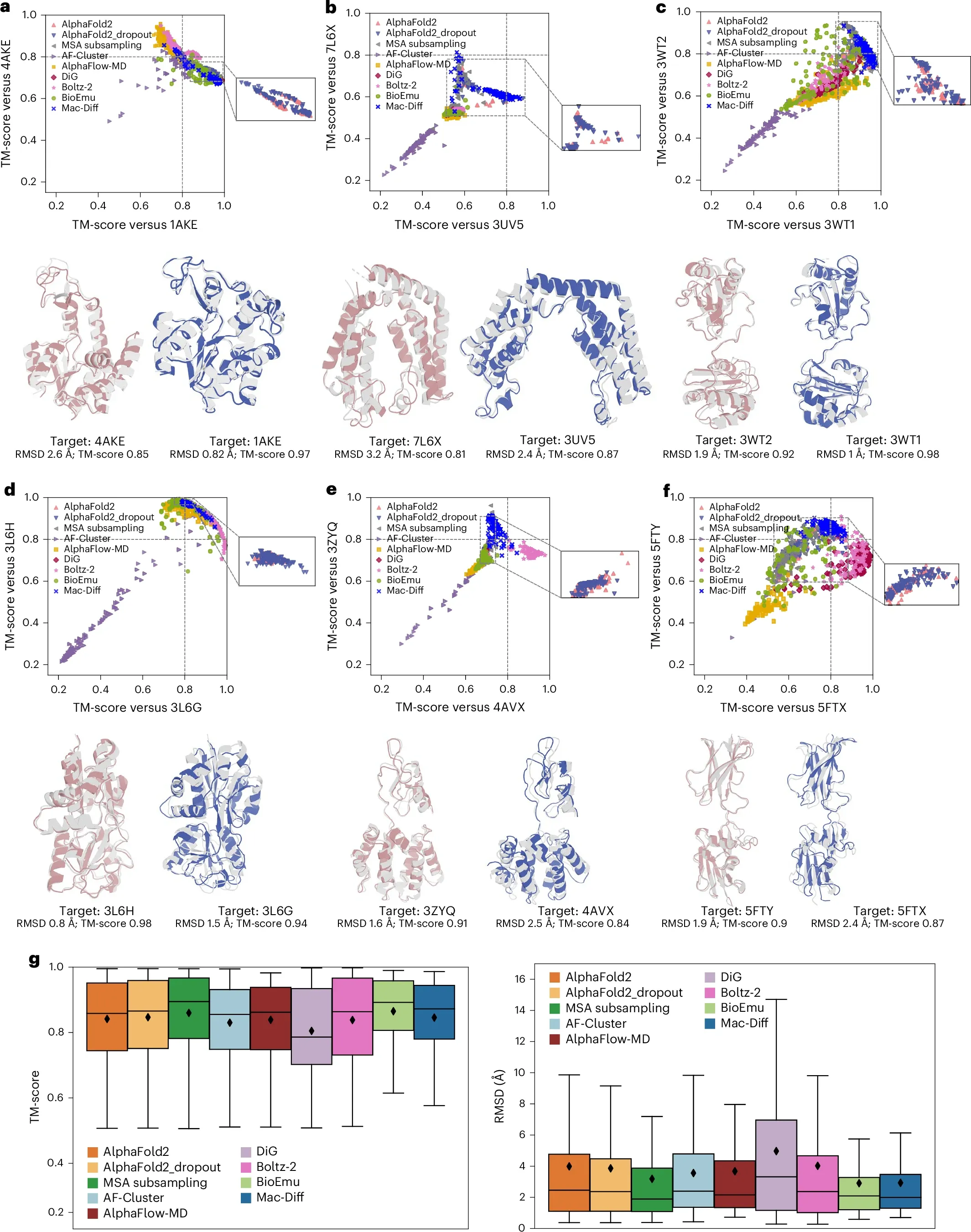
图6 Mac-Diff和8种对比方法在变构蛋白AdK及Cfold40测试集中代表案例上的表现。(a-f) 模型在Adk、TAF1的串联溴结构域、PDI的的b’–a’域、OpuA的底物结合域、HGS-Hrs的串联VHS和FYVE结构域及表面层蛋白上性能。(g) 9种方法的TM-score和RMSD分布[1]
总结
综上所述,Mac-Diff通过引入局部感知的模态对齐注意力机制,既保留了进化驱动的序列表征的丰富信息,又融入了物理先验的几何约束规律。其不仅在蛋白质平衡构象生成的准确性与多样性上取得了显著突破,还展现了捕捉关键功能构象的卓越能力,为变构药物设计及蛋白质动力学机制的深入解析提供了全新思路和强有力的技术支撑。
王保利、王成林、陈劲杨为本研究的共同第一学生作者,参与本研究工作的还有华东师范大学药学院/人工智能新药创智中心刘旦麟副研究员、孙长志博士等。该工作得到了中国国家自然科学基金 (82425104, 62276099) 和国家重点研发计划 (2022YFC3400501) 的资助。
参考文献
[1] Wang, B., Wang, C., Chen, J. et al. Conditional diffusion with locality-aware modal alignment for generating diverse protein conformational ensembles. Nat Mach Intell (2026).
原文链接:https://www.nature.com/articles/s42256-026-01198-9
供稿:王保利
编辑:汤荣凡
华东师范大学药学院/人工智能新药创智中心/李洪林教授课题组
文章改编转载自微信公众号:DrugOne
原文链接:https://mp.weixin.qq.com/s/jnI6G6bZ95gRPgTK6n58tQ?scene=1 | 





















