本文解读 ACS Nano 2025 年论文《A Machine Learning Framework for Modeling Ensemble Properties of Atomically Disordered Materials》,该研究提出等变图神经网络 + 蒙特卡洛统一框架,以 Ti₃C₂Tₓ MXene 为模型体系,毫秒级预测能量、光 / 电导率谱,精准揭示有序 - 无序相变规律;发现电导率在相变温度附近出现特征峰,光导率对局部无序不敏感,仅由表面化学组成决定,为高熵合金、自旋液体等无序材料建模提供通用方案。

在二维材料的大家族里,MXene 凭借优异导电性、可调表面化学和出色机械性能,成为储能、催化、电磁屏蔽领域的明星材料。但它有一个 “天生的麻烦”:表面原子高度无序。-O、-F、羟基、空位随机分布,让每一块 MXene 的原子排布都不一样,而实验测到的都是 “平均后的结果”。传统第一性原理算不动、统计方法算不准,长期以来,没人能高效把 “局部原子无序” 和 “宏观光电性能” 精准连接起来。直到这篇发表在ACS Nano的工作出现 —— 科学家用图神经网络 + 蒙特卡洛模拟,造出一套专门破解 “原子无序材料系综特性” 的 AI 计算框架,把无序 MXene 的热力学、电导、光导行为算得明明白白,速度和精度双双拉满。
01 原子无序:材料科学的 “迷雾难题”
原子无序是材料中普遍存在的现象,只要原子排列偏离理想晶格,就可以归为无序结构,主要包括构型无序与结构无序两大类。构型无序指不同原子、官能团或自旋态在晶格位点上随机分布,而结构无序则以空位、位错等破坏化学键的形式存在。这类局部的原子排布混乱,看似细微,却能彻底改变材料的导电、导热、光学响应与催化活性,是高熵合金、热电材料、非晶固体等体系性能调控的核心因素。
然而,想要准确模拟无序材料,传统计算方法面临着难以突破的瓶颈。首先,第一性原理密度泛函理论(DFT)计算成本极高,单个构型的光谱与输运性能往往需要数小时,无法支撑成千上万种构型的统计采样。其次,实验测量的是宏观系综平均结果,而传统计算多基于单一理想结构,难以与实验直接对照。最后,团簇展开、分子动力学等加速方法虽然能提升结构采样效率,却很难准确预测光导率、电导谱这类由电子结构主导的复杂谱学性能。可以说,原子无序长期笼罩在计算材料学的迷雾之中。
02 AI 破局:GNN + 蒙特卡洛全能计算框架
2.1 框架整体设计
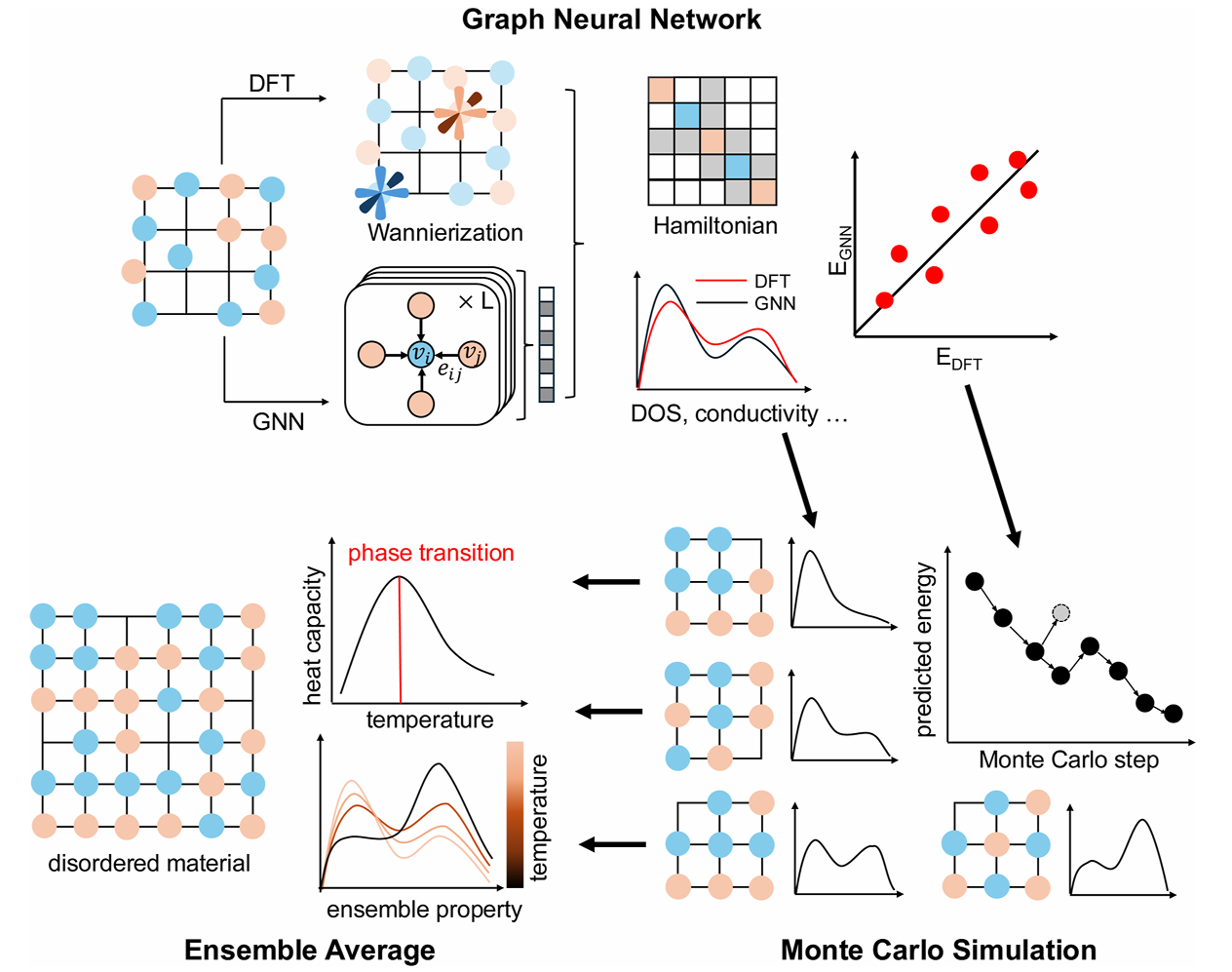
图1 面向原子无序材料的GNN+MC机器学习计算框架
为了攻克无序材料的计算难题,这项研究搭建了一套融合等变图神经网络(GNN)*与*蒙特卡洛(MC)模拟的通用机器学习框架,实现了从原子结构到热力学、光电性能的全流程高效预测。这套框架的核心思路非常清晰:先用高精度数据集训练神经网络,让 AI 学会原子结构与性能之间的映射关系,再将训练好的模型嵌入蒙特卡洛模拟,快速采样海量无序构型,最终统计得到可与实验对比的系综平均性能。
整个流程高度自动化:先通过高通量 Wannier 函数流程生成训练数据,再训练 GNN 模型,最后将模型嵌入 MC 模拟进行大规模采样与统计平均,兼顾了物理对称性、预测精度和计算效率。
2.2 模型关键改进:让 AI 看懂空位与无序
普通 GNN 很难识别空位这类 “没有原子” 的结构,而无序 MXene 最关键的结构特征恰恰是端基空位与官能团乱序。为此,研究团队对模型进行了两项关键改进:
(1)在节点特征中加入持久同调拓扑特征,让模型自动捕捉空位数量、位置与局部成键环境;
(2)将空位位点设为虚拟节点直接参与图网络运算,强化模型对缺陷与无序的感知能力。
这一设计让模型对无序结构极度敏感,完美适配氧化物、二维材料、金属玻璃等各类无序体系。
2.3 核心公式:热力学平均与性能统计
为了定量描述无序体系的热力学与输运行为,研究中使用了两个核心公式,也是整个框架的物理基础。
(1)有序 - 无序相变判定:定容热容
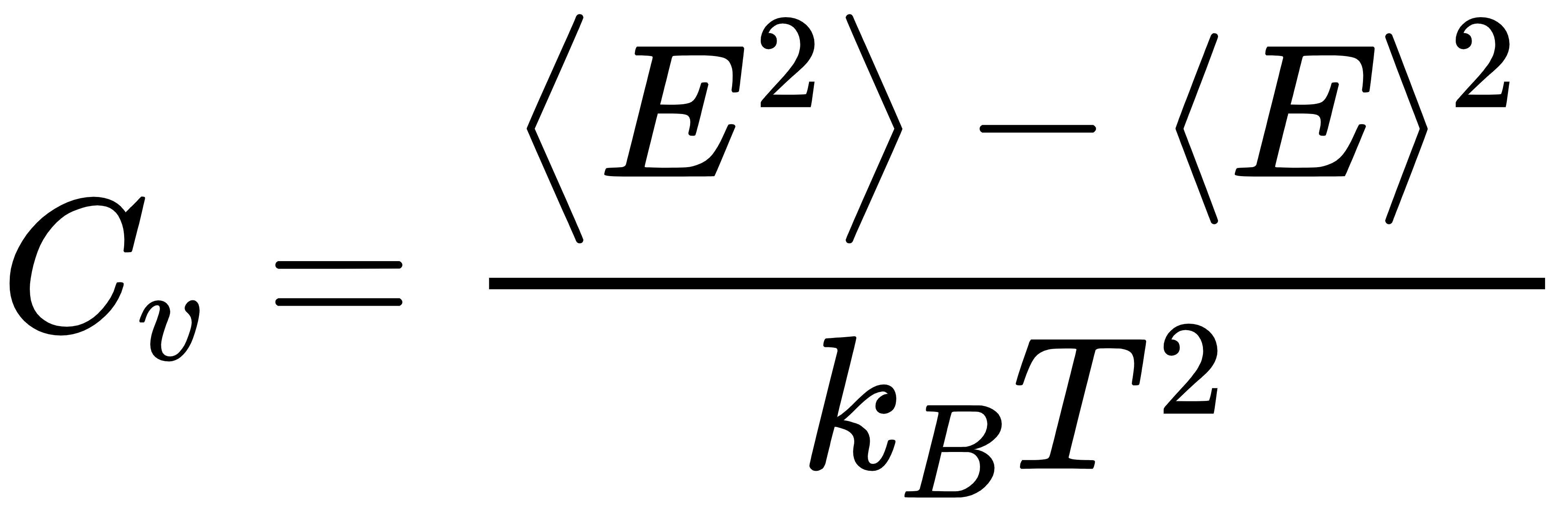
热容出现峰值的温度,就是材料发生有序 - 无序相变的临界温度。
(2)系综平均电导率(调和平均)
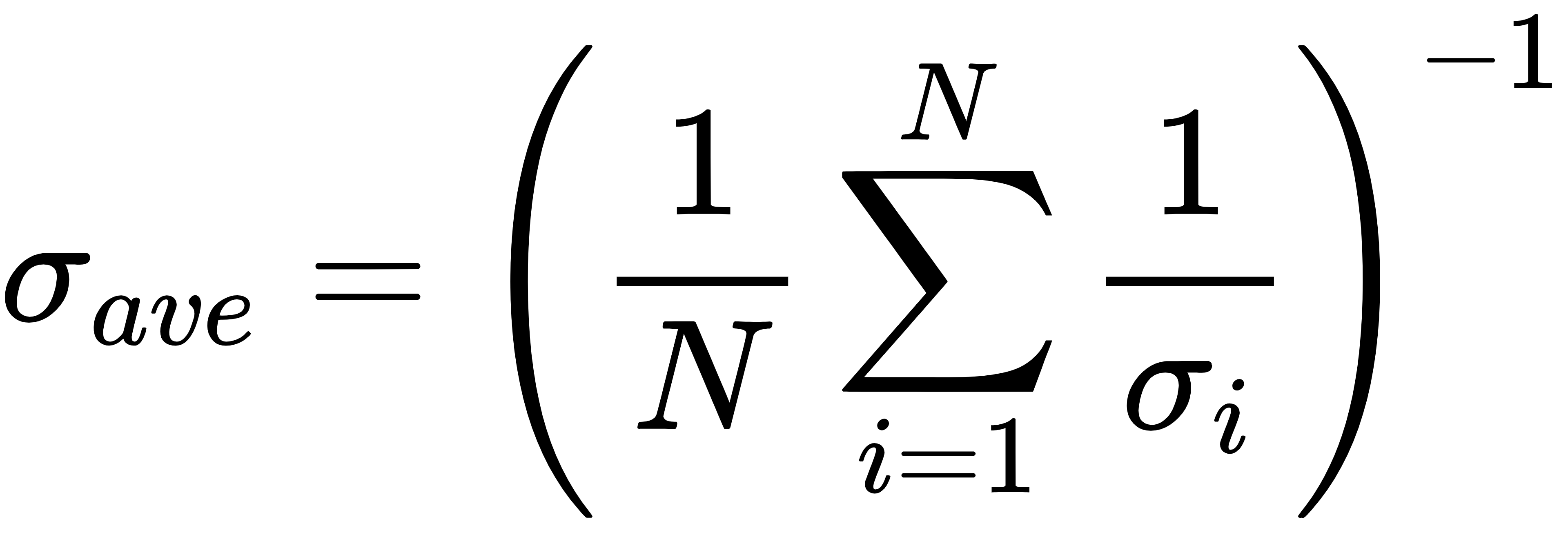
更符合无序体系 “串联导电” 的真实物理图像,能给出更接近实验的宏观电导。
03 关键结果:精准还原无序 MXene 的光电与相变行为
3.1 AI 预测精度:逼近 DFT,速度提升万倍
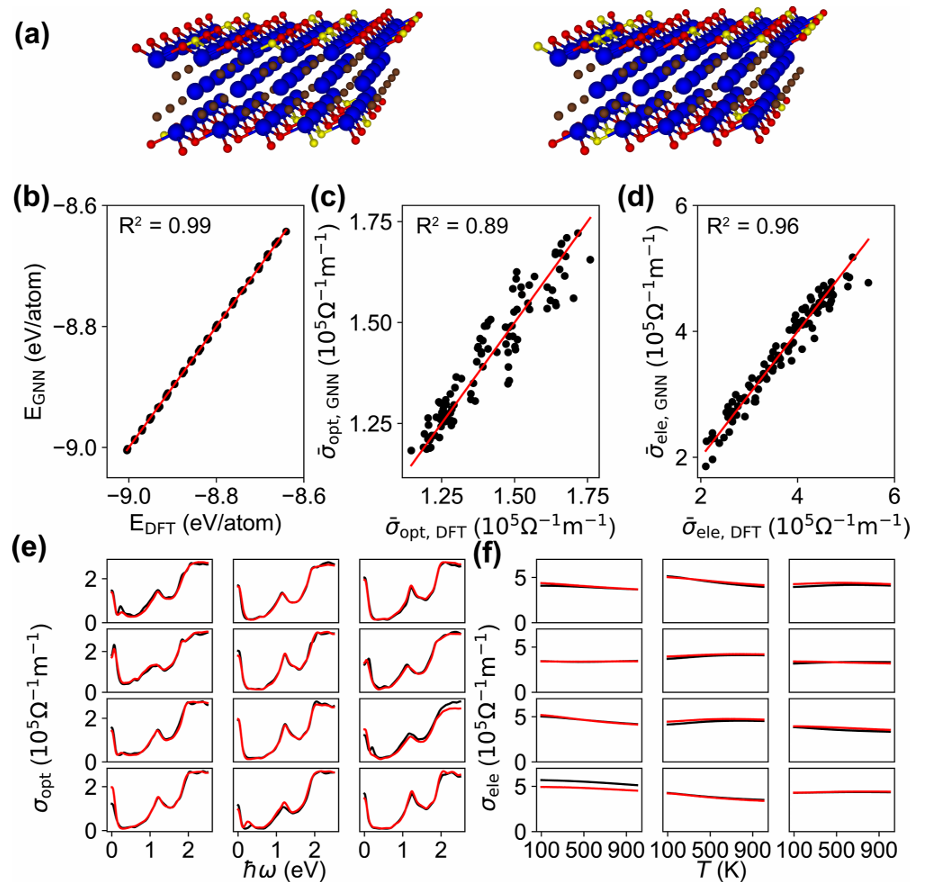
图2 GNN预测与DFT计算结果
经过系统训练,该模型同时实现了能量、光导率、电导率三类关键物性的高精度预测。其中能量预测决定系数R²=0.99,平均相对误差仅0.02%;电导率预测R²=0.96,光导率预测R²=0.89,整体输出与 DFT 第一性原理结果几乎完全重合。更具应用价值的是,模型完成单个构型预测仅需毫秒级时间,相比传统 DFT 计算速度提升上万倍,使得大规模无序构型采样与统计成为可能。
3.2 热力学规律:F 与空位抬高清有序 - 无序相变温度
依托训练好的模型与蒙特卡洛采样,研究首次定量揭示了 MXene 表面无序的热力学规律。随着表面 -F 官能团比例升高以及空位浓度增加 ,体系的有序 - 无序相变温度会显著提高。这是因为氟原子与空位会显著提高结构进入完全无序状态的能量势垒,让局部原子排列更难被打乱,从而在更宽温度范围内保持局部有序特征。
3.3 电导率行为:相变温度附近出现特征峰
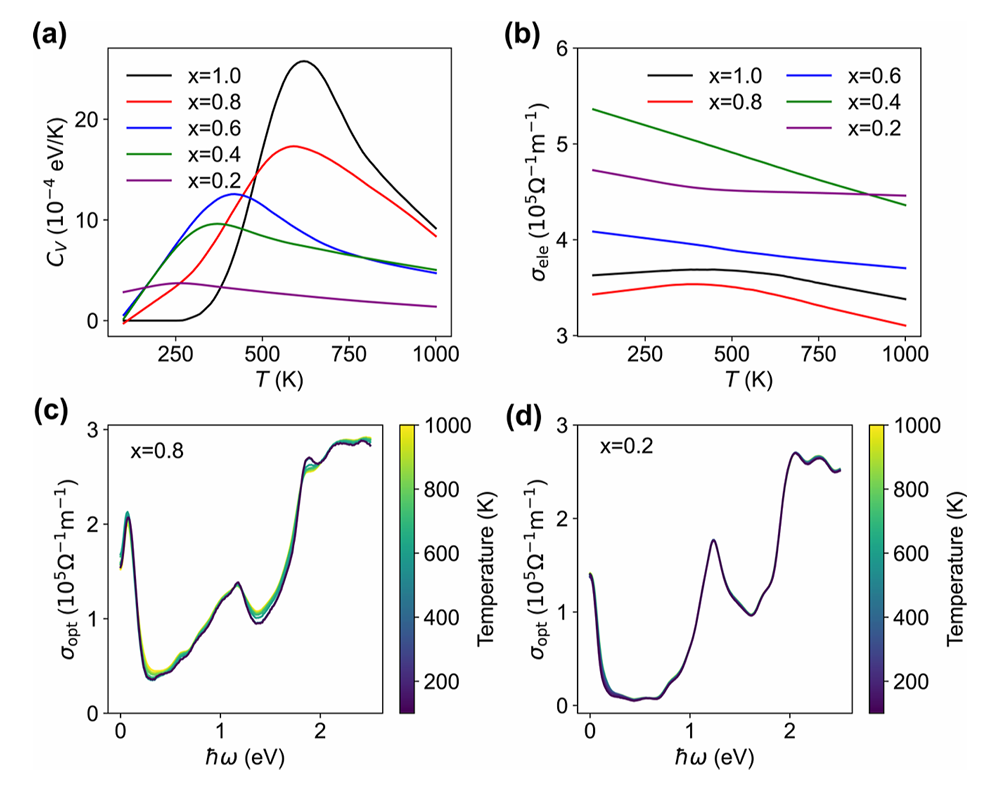
图3 有序-无序相变及系综平均光电性能
在电子输运性能上,电导率表现出对原子无序的高度敏感性。在低氟含量时,电导率随温度升高单调下降;而在高氟含量下,电导率会在有序 - 无序相变温度附近出现明显特征峰。这一现象源于电子散射与载流子掺杂的竞争效应:低温下 F 原子充当散射中心,高温下则表现为电子掺杂,共同导致了电导峰的出现。同时,表面空位会显著抑制面内电荷输运,进一步降低电导率,与实验观测高度一致。
3.4 光导率行为:对无序不敏感,仅由化学组分决定
与电导率形成鲜明对比,光导率展现出极强的无序鲁棒性。在不同温度、不同无序程度下,光导率谱形几乎保持不变,其在1.5 eV 处的特征峰仅由表面整体化学组分决定,-F 比例越高,峰强度越弱。这是因为光响应主要来自带间跃迁,基本不受载流子散射与局部原子排布的影响。这一规律也为实验表征提供了新思路:用光导率测成分,用电导率测无序。
3.5 含空位真实体系:泛化能力与实验场景一致
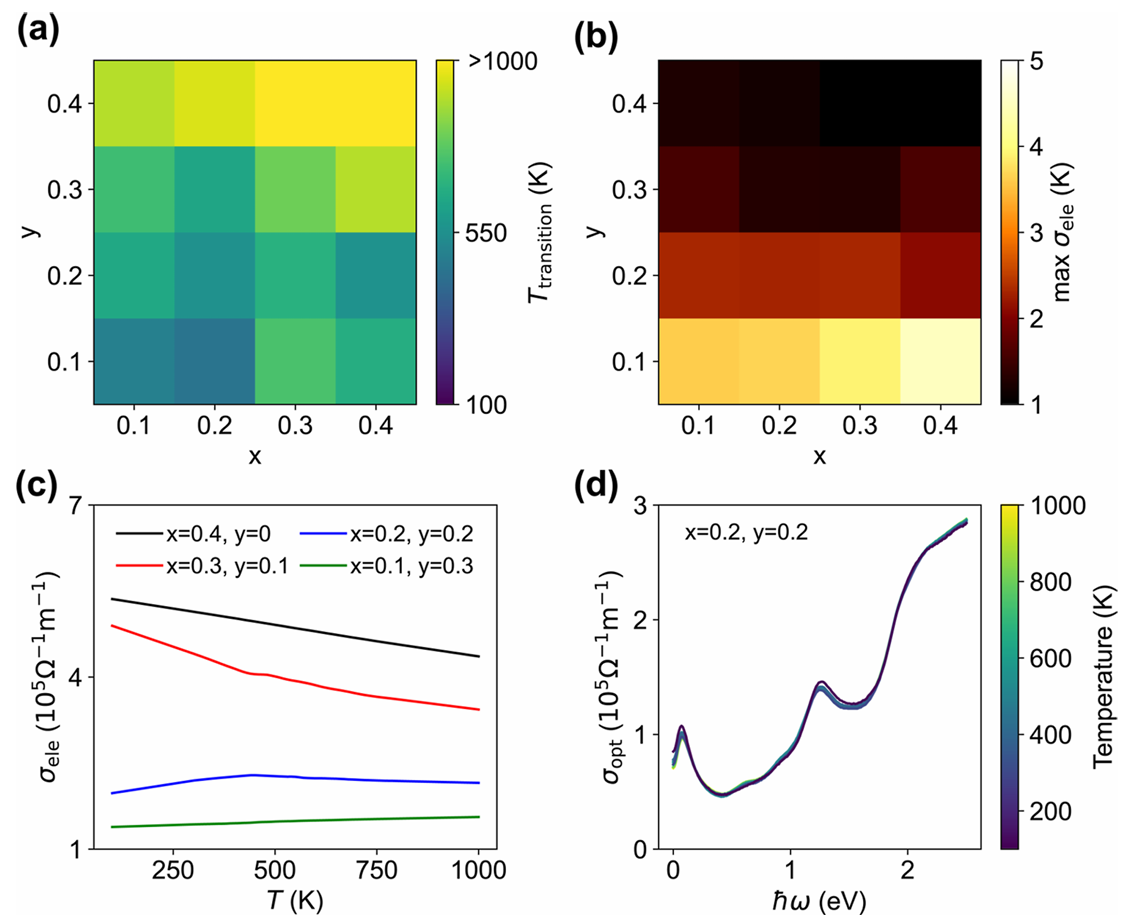
图4 含空位无序MXene的热力学与光电响应
为贴近真实 MXene 材料,研究进一步构建了包含端基空位的 Ti₃C₂O₂₋ₓ₋ᵧFₓ数据集。模型在该体系下依然保持超高精度,能够准确预测空位对相变温度、电导率的调控作用,并正确区分不同环境下的空位形成能力:气相退火条件下空位难以形成,而水溶液、电化学环境下可出现稳定空位,完全符合实际材料的制备与服役场景。
04 科学意义:无序材料研究进入 AI 时代
这项工作的价值,不仅在于解决了 MXene 表面无序的计算难题,更在于建立了一套通用、高效、高保真的无序材料研究范式。它首次实现了电子谱学性能与热力学性质的同步系综平均计算,首次用 AI 定量揭示了无序对二维材料光电性能的调控机制,让过去无法完成的大规模无序构效关系研究变成常规操作。
这套框架并不局限于 MXene 体系,具备极强的可迁移性,未来可直接用于高熵合金、非晶材料、热电材料、自旋液体等复杂体系的无序效应研究,也可以与实验电子断层扫描数据结合,实现结构解析与性能预测的一体化。它真正证明,人工智能不再是简单的拟合工具,而是可以直接揭示物理规律、驱动材料发现的核心引擎。
05 总结
原子无序是材料科学中无法回避却长期难以攻克的课题,它既是影响性能的关键因素,也是计算模拟的巨大障碍。这篇发表于 ACS Nano 的研究,用等变图神经网络与蒙特卡洛模拟的强强联合,为无序材料打开了全新的研究局面。它以超高速度、超高精度同时还原结构、热力学、电导、光导与相变行为,清晰区分了成分与无序对 MXene 性能的不同作用,为二维材料的理性设计提供了可靠工具。随着这套 AI 框架的不断拓展,高熵合金、非晶固体、能源催化等领域的无序之谜,将被逐一解开。
论文链接:https://pubs.acs.org/doi/10.1021/acsnano.5c13080 | 





















