本文解读 arXiv 预印本论文《Chemistry-Enhanced Diffusion-Based Framework for Small-to-Large Molecular Conformation Generation》,研究提出StoL(Small-to-Large)框架,将化学先验与扩散模型结合,采用 “LEGO 式片段组装” 策略,仅用小分子数据训练,即可直接生成大分子高质量 3D 构象。该方法无需大分子训练集、不依赖外部软件,在药物分子上实现比传统工具更全面的构象覆盖,且能找到能量更低的稳定结构,为药物设计、催化模拟提供轻量高效的构象生成方案。

分子的 3D 形状,直接决定它能不能成药、稳不稳定、怎么反应。可越大越灵活的分子,构象数量就指数爆炸,想算出一套靠谱的 3D 结构,要么慢到离谱,要么不准到没法用。传统方法要么靠量子化学暴力搜索,算力烧不起;要么靠经验规则快速生成,却容易漏掉关键低能构象。
最近,一种叫StoL的新框架彻底换了思路:把大分子拆成碎片,用 AI 生成碎片,再像拼乐高一样搭回大分子。更厉害的是,它只学过小分子,就能直接造大分子的 3D 构象,还自带化学规律约束,生成又快又稳。
一、大分子 3D 构象:计算化学的 “老大难”
想要准确预测分子性质,第一步就是拿到合理的 3D 构象。但分子越大、可旋转单键越多,构象空间就越恐怖。
传统的分子动力学、元动力学虽然精度高,但对大分子做全空间搜索成本极高,很难把低能构象找全。而基于经验规则的快速工具,虽然快,却常常局限在局部构象区间,覆盖不够全面,也容易出现不合理的键长、键角和原子碰撞。
近几年火起来的扩散模型,给分子生成带来新希望,但多数模型都是纯数据驱动,缺少化学规律约束,很容易生成不符合化学常识的结构,而且必须用大量大分子数据训练,对没多少数据的新分子根本不友好。
科研界一直想要一个两全其美的方案:够快、够准、够化学合理,还不用啃大分子数据集。
StoL 的出现,正好补上了这块缺口。
二、StoL:用 “小分子碎片” 拼出 “大分子构象”
StoL 的全称是 Small-to-Large,核心逻辑非常直观:不直接生成大分子,而是拆成碎片生成,再拼回去。
2.1 整体流程:拆解 — 生成 — 组装三步法
整个流程像极了用乐高搭模型,清晰又好理解:
片段拆解:把目标分子按化学规则切成合理小碎片,只切环外单键,保证每块碎片化学上合法。
碎片生成:用训练好的扩散模型,给每块碎片生成一批高质量 3D 构象。
全局组装:按重叠区对齐,把碎片一块块拼回完整分子,再做化学校验,去掉不合理结构。
最惊艳的是,StoL全程不用任何大分子结构训练,只需要小分子碎片数据,就能轻松扩展到大分子,可迁移性极强。
2.2 化学增强扩散模型:让 AI 懂化学常识
普通扩散模型只学数据,StoL 则把化学规则 “焊” 进模型里,训练分两步:
1.先做常规数据驱动训练,搭好分子骨架;
2.再进入化学增强阶段,处理对称、原子匹配、芳香环平面性。
它用 Sinkhorn 算法处理原子等价性,用 Gumbel-softmax 稳定原子序,还专门加平面性约束,不让芳香环歪掉。这样训练出来的模型,收敛更快、结构更合理,不会出现物理上荒唐的构象。
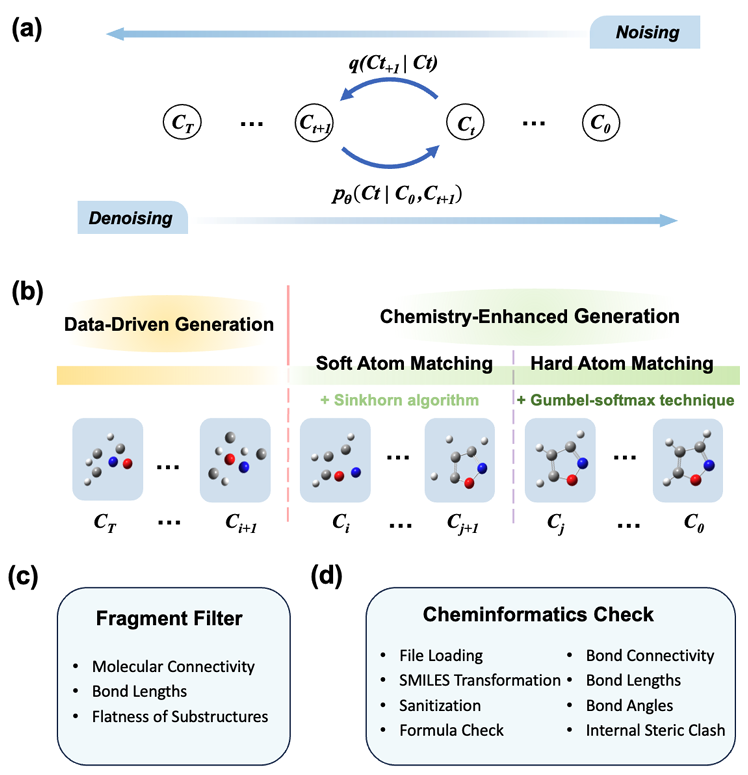
图1 化学增强扩散模型训练与结构验证流程
2.3 组装与过滤:只保留化学合法结构
拼碎片不是硬凑,而是通过共享原子区域精确对齐,只保留 RMSD 足够小的组合。组装完成后,还会做一套完整化学检查:键长、键角、连通性、原子冲突、分子式一致性……
层层过滤后,输出的构象直接可用,不用再手工修结构。
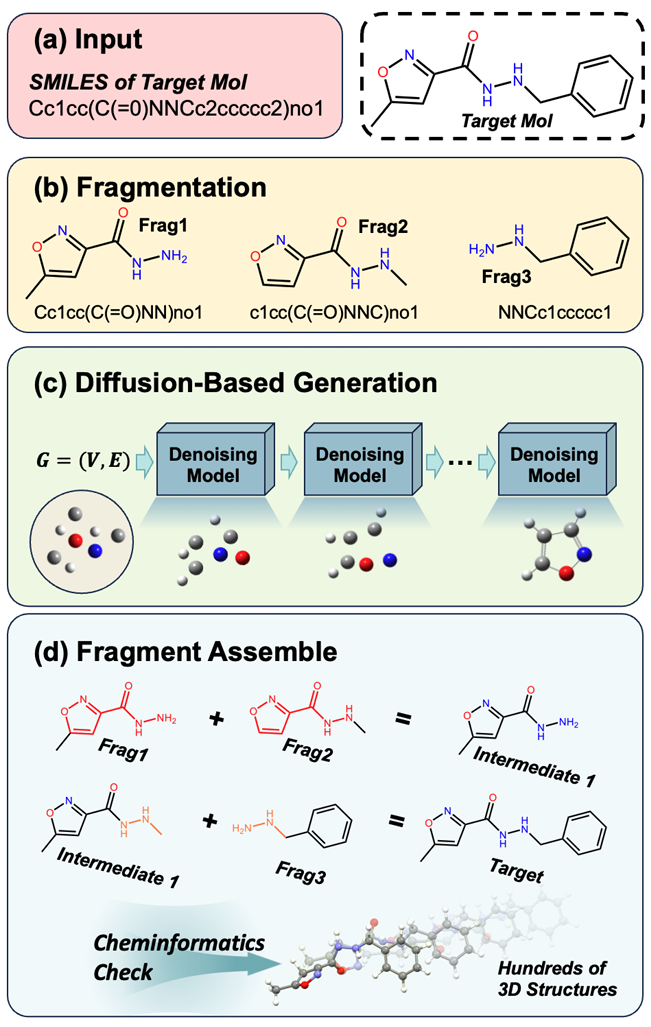
图2 StoL 框架:从 SMILES 输入到 3D 分子构象的全流程
三、真实效果:药物分子测试完胜传统工具
研究团队选择了抗癌药物伏立诺他(Vorinostat) 做典型演示,这分子长且柔性高,是公认的构象生成难题。
StoL 先把它切成三块,分别生成碎片构象,再组装成完整分子。结果显示,StoL 生成的构象在空间里分布更广、更均匀,而传统工具 RDKit 只集中在一小块区域。
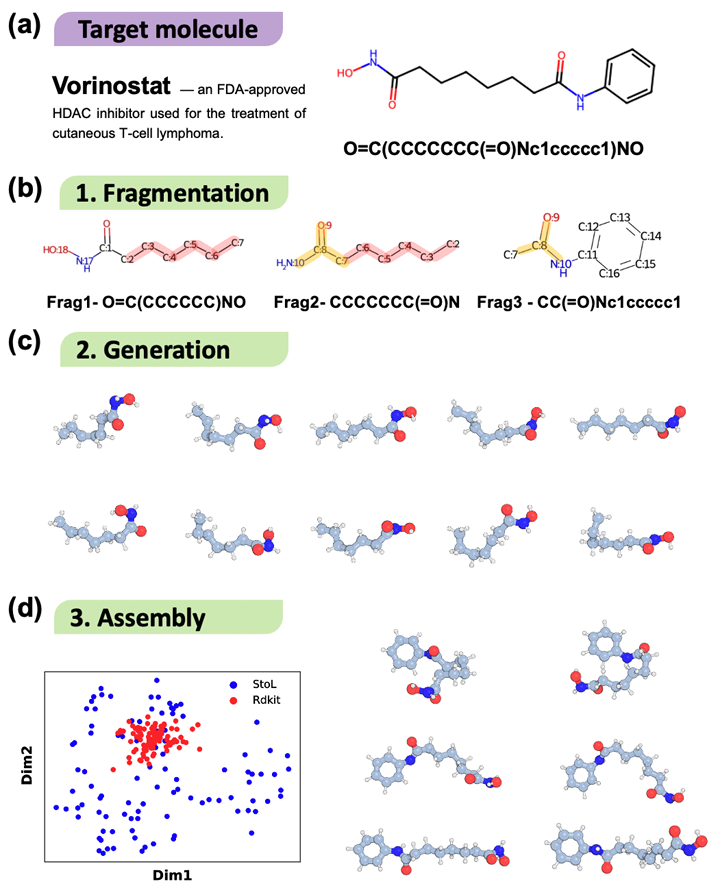
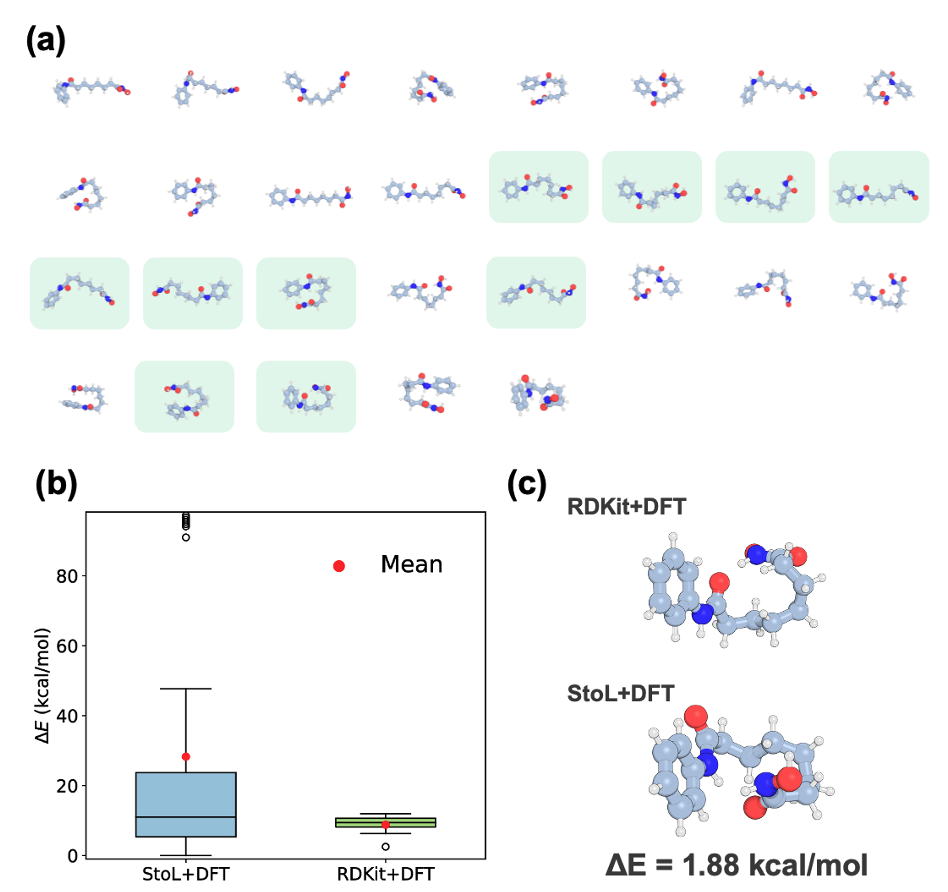
图3 药物分子 Vorinostat 构象生成与能量优化对比
经过 DFT 量子化学优化后,对比更加明显:
1.StoL 能覆盖 RDKit 找到的所有构象,还能找到更多新构象;
2.找到的最低能构象,比传统方法低出1.88 kcal/mol;
3.构象能量区间更宽,更接近真实势能面分布。
在包含 200 个类药分子的 StoL25-init 测试集上,StoL 同样表现稳定:
超过一半分子的玻尔兹曼加权 RMSD 低于 1 Å,整体几何精度极高;65% 的分子能找到比传统方法更低的能量构象,真正做到 “构象找得全、低能找得准”。
四、碎片策略重新定义分子构象生成
StoL 最核心的突破,是用 “从小到大” 的片段化思路,绕开了大分子生成最头疼的数据瓶颈。它不再需要庞大的大分子训练集,只依靠小分子片段数据,就能实现高质量、高覆盖的构象生成,让大体系分子模拟不再被算力和数据卡脖子。
将化学先验嵌入扩散模型的训练与组装全过程,不仅让结构更合理、训练更稳定,也让整个流程端到端、不依赖外部软件,简单易用。这种 “物理规则 + AI 生成” 的模式,为下一代分子生成模型提供了可复制的范式。
相比于传统方法,StoL 在构象覆盖度、结构合理性、低能构象搜索能力上全面占优,尤其适合柔性药物分子、环境污染物、有机催化中间体等缺乏高质量构象的体系。
五、总结
分子越大,构象越难算,这是计算化学持续多年的痛点。StoL 用 “拆解 — 生成 — 组装” 的 LEGO 式策略,配合化学增强扩散模型,给出了一套轻巧却强大的解决方案。它只学小分子,就能做大分子;只输 SMILES,就能出一整套合理 3D 构象;比传统工具覆盖更广,比纯数据 AI 更化学可信,还能稳定找到能量更低的优势构象。从药物筛选、分子模拟到催化设计,StoL 都能快速提供高质量初始构象,把过去要算几天的构象搜索,压缩到小时甚至分钟级。
这是一次真正 “又快又好又省” 的突破,让大分子 3D 构象生成,从此走出算力与精度的两难困境。
论文链接:https://arxiv.org/abs/2511.12182v1 | 





















