本帖最后由 宇宙微尘 于 2026-5-18 11:40 编辑
本文解读《Nature Machine Intelligence 2026》成果《Force-free molecular dynamics through autoregressive equivariant networks》,一种基于自回归等变消息传递网络的无力分子动力学(Force-free MD)全新框架。它彻底抛弃传统分子动力学(MD)必需的原子间作用力计算与数值积分步骤,直接由当前时刻原子位置、速度预测下一时刻运动状态,将模拟时间步长扩大10–30倍,单日可生成最高15 ns的原子轨迹。TrajCast在扑热息痛小分子、α-石英晶体、液态水等多体系中精准复现结构特征、动力学行为与能量分布,更实现零样本泛化至超淬冷水玻璃化这一经典难题。文末将简要介绍玻色量子相干光计算如何为其赋能,进一步突破经典模拟极限,为材料、化学、生物医药等领域的微观研究带来范式革新。

在现代科学研究中,分子动力学模拟(MD)就像一台“数字显微镜”,能让我们在原子尺度“亲眼看到”微观世界的运动:蛋白质如何折叠成特定结构、晶体如何发生相变、催化剂表面如何发生反应、液态水的分子如何快速运动……这些实验难以直接观测的过程,都能通过MD模拟清晰呈现。它不仅是基础科研的核心工具,更支撑着新药研发、新材料设计、新能源器件开发等多个产业的发展。
但这台“数字显微镜”却长期被一个致命问题困扰:运行效率极低。传统MD的核心逻辑几十年来从未改变,始终遵循“算力→积分→更新”的固定循环:第一步,计算体系中每一个原子受到的所有作用力(包括成键作用、范德华作用、库仑作用等);第二步,通过数值积分求解牛顿运动方程,将作用力转化为原子的位移和速度变化;第三步,更新所有原子的位置和速度,重复这三个步骤,直到获得足够长的轨迹。
为了保证模拟的热力学稳定性和能量守恒,时间步长必须被严格限制在0.5–1 fs(飞秒,1 fs = 10⁻¹⁵秒)的极小范围。这意味着,要模拟1 ns(纳秒,1 ns = 10⁻⁹秒)的原子运动,需要推进超过100万步;如果要模拟玻璃化、蛋白质折叠这类慢动力学过程(通常需要微秒甚至毫秒级轨迹),即便使用最先进的超级计算机和机器学习势函数,也需要耗费数月甚至数年的时间。长期以来,“必须算力、必须积分、步长极小”成为悬在原子模拟领域上方的三座大山,让很多前沿课题难以高效开展。
而TrajCast的出现,彻底打破了这一僵局。它提出了一种颠覆性的“无力”设计——完全不计算原子间作用力,不进行数值积分,直接用AI模型一步预测原子的下一步运动状态。这种设计从底层重构了分子动力学的运行逻辑,让模拟效率实现了数量级的提升,同时还能保持极高的精度,开启了原子模拟的第三代范式。
一、传统分子动力学的底层瓶颈:为什么“不算力就动不了”?
要理解TrajCast的革命性,首先要搞清楚传统MD的核心瓶颈到底在哪里。传统MD的本质是“基于力的动力学演化”,这种模式存在三个无法绕过的固有瓶颈:
第一,力计算成本极高。无论是基于第一性原理(如密度泛函理论DFT)还是机器学习势函数(MLIP),计算每一个原子的受力都需要遍历其周围的原子,进行复杂的电子结构计算或特征拟合。对于包含数千、数万个原子的大体系,每一步的力计算都需要消耗大量算力。
第二,数值积分限制步长。原子间的化学键振动频率极高(约10¹³–10¹⁴ Hz),对应的特征时间约为10 fs。根据数值积分的稳定性要求,步长必须远小于这一特征时间,因此传统MD的步长只能限制在0.5–1 fs。这就导致长时程模拟需要推进海量步骤,效率极低。
第三,误差累积导致轨迹失真。数值积分本身存在截断误差,每一步的微小误差会随着步数的增加不断累积,长期模拟后,体系会偏离真实的热力学系综,原子结构和动力学行为与实际情况产生偏差,影响模拟结果的可靠性。
多年来,科研人员尝试过多时间步积分(对高频振动用小步长,低频运动用大步长)、GPU并行加速、高通量采样等方案,但这些都只是“治标不治本”,始终无法跳出“先算力、再积分”的底层框架。而TrajCast的革命性,就在于它完全跳过了“力”这一中间变量,直接构建从当前原子状态到下一状态的映射关系,从根源上解决了效率瓶颈。
二、TrajCast核心原理:自回归等变网络,真正实现“无力”动力学
TrajCast的全称是Trajectory Forecaster,直译是“轨迹预测器”,它的核心定位是:一种基于自回归序列预测与E(3)等变图神经网络的原子轨迹预测模型。其核心功能非常简单:输入t时刻的原子位置(r(t))、速度(v(t))与元素类型(Z),直接输出t+Δt时刻的位置(r(t+Δt))与速度(v(t+Δt)),全程不计算力、不进行数值积分。
要让AI模型输出的结果符合物理规律,TrajCast在架构设计中嵌入了三类严格的物理约束,这也是它区别于普通预测模型的关键,更是其能够精准复现原子动力学的核心原因。此外,论文中引入玻尔兹曼分布作为热力学约束,进一步保证了模拟结果的物理合理性,这也是TrajCast精准度突出的重要支撑。
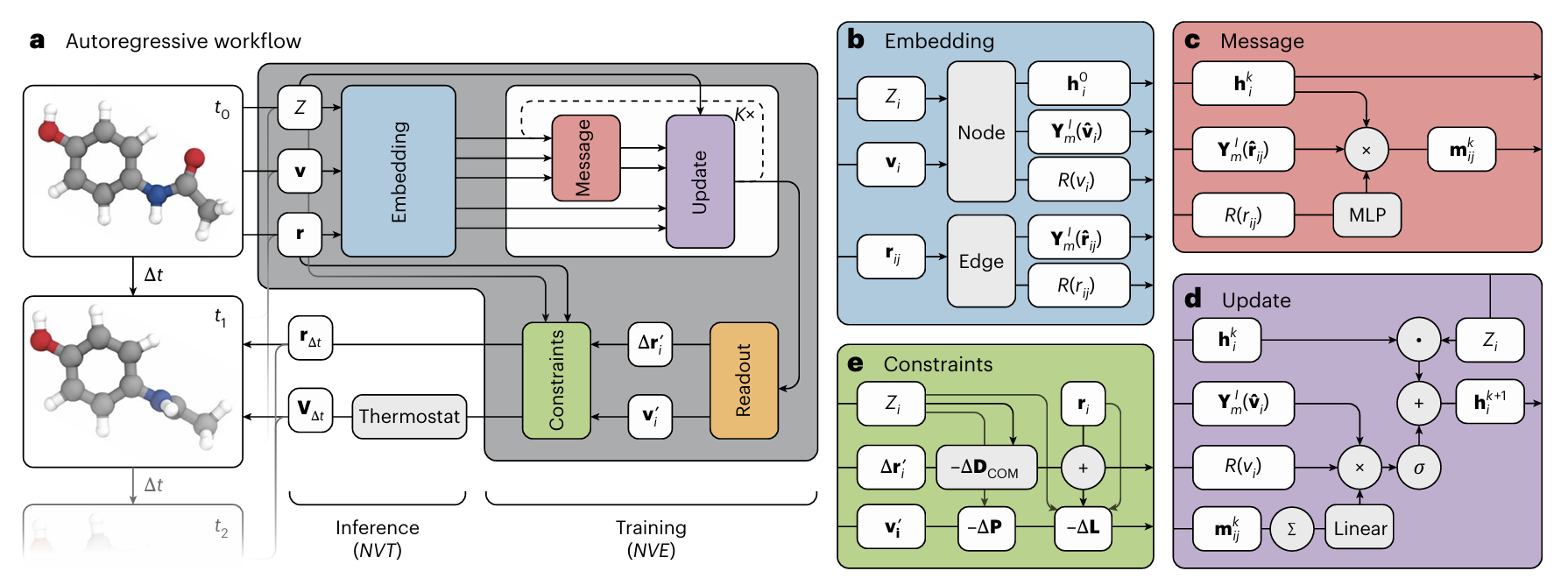
2.1 核心约束1:E(3)等变,保证几何对称性
原子系统具有旋转、平移、反演不变性——也就是说,无论整个体系如何旋转、平移,原子间的相对作用关系和运动规律都不会改变。如果模型不满足这种对称性,预测结果就会偏离物理 reality。TrajCast采用了E(3)等变图神经网络(Equivariant Graph Neural Network, EGNN),确保模型的输出会随着输入的旋转、平移而同步变化,严格遵守原子系统的几何对称性。
这种等变性通过“等变消息传递”实现:
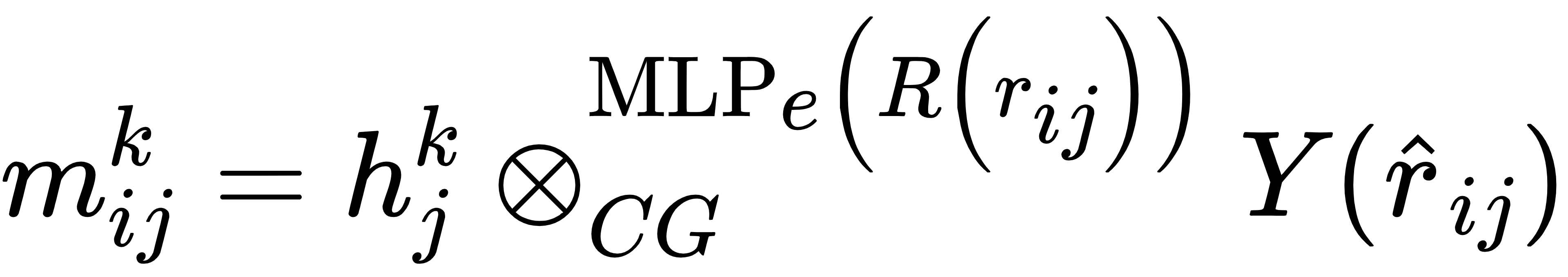
其中,mijk是原子i从原子j接收的第k阶消息,hjk是原子j的第k阶特征,⊗CG是Clebsch-Gordan张量积,MLPe(R(rij)) 是基于原子间距离 rij的多层感知机,Y(r^ij) 是球谐函数。
简单来说,这个公式的作用是:在传递原子间信息时,不仅考虑原子间的距离,还考虑相对方向,同时通过Clebsch-Gordan张量积保证,无论体系如何旋转、平移,信息传递的结果始终符合物理规律,让模型真正“理解”原子的局部环境。
2.2 核心约束2:显式建模速度,覆盖完整相空间
原子的运动状态不仅包括位置,还包括速度——位置描述“在哪里”,速度描述“往哪动、动多快”,两者共同构成原子的相空间。传统MD中,速度是通过力计算和积分得到的,而TrajCast直接将速度作为输入和输出的一部分,显式建模速度的演化规律,确保模型能够捕捉到原子运动的完整信息。
模型的输入是一个联合特征向量:xi =[ri,vi, Zi],其中Zi是原子i的元素类型(用于区分不同原子的特性)。输出则是t+Δt时刻的位置偏移量Δri和新速度 vi,最终通过以下公式得到下一时刻的位置:
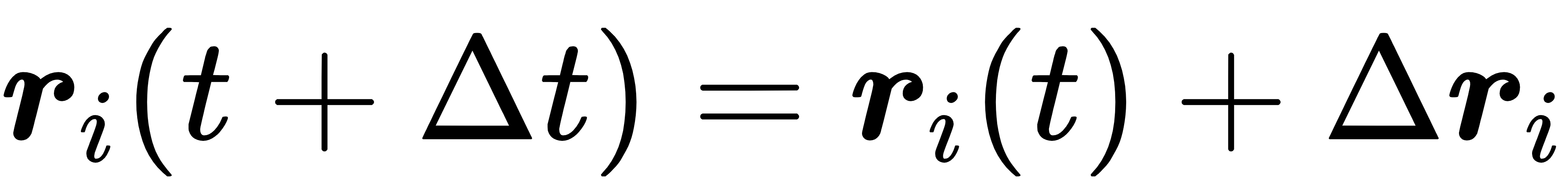
这种设计避免了仅预测位置带来的热力学信息缺失,让模型生成的轨迹能够稳定对应真实的热力学系综(如NVE系综:粒子数、体积、能量守恒;NVT系综:粒子数、体积、温度守恒)。
2.3 核心约束3:内置守恒律,保证轨迹稳定
经典力学中,线动量和角动量守恒是原子系统必须遵守的基本规律。如果模型预测的轨迹不满足守恒律,就会出现“能量不守恒”“体系自发旋转”等不符合物理现实的情况。TrajCast内置了线动量和角动量修正模块,在每一步预测后,对原子的位置和速度进行微调,确保整个体系的线动量和角动量始终守恒。
线动量守恒修正的核心公式的是:
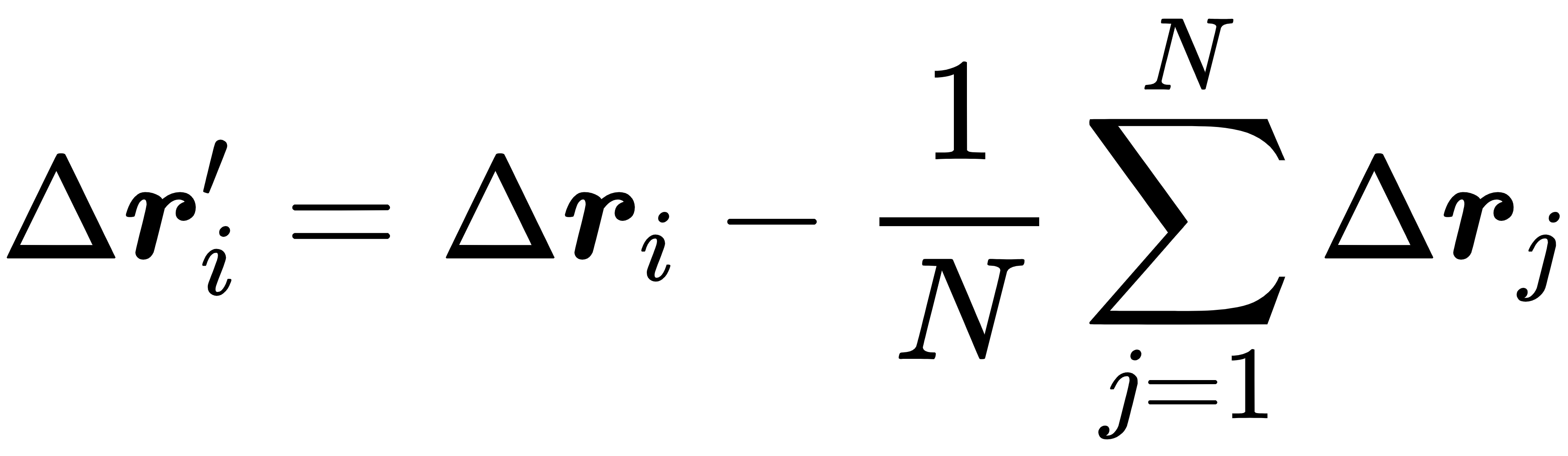
其中,N是体系中的原子总数,Δri'是修正后的位置偏移量。通过这种方式,消除体系整体的平移偏差,保证线动量守恒。角动量守恒修正则通过调整原子的速度方向实现,确保体系不会出现自发的旋转。
2.4 自回归滚动预测:实现长时程轨迹生成
TrajCast采用自回归(Autoregressive)预测模式,即:将t时刻的预测结果作为t+Δt时刻的输入,持续滚动预测,从而生成连续、稳定的长时程原子轨迹。其整体预测逻辑可以用一个统一的映射公式表示:
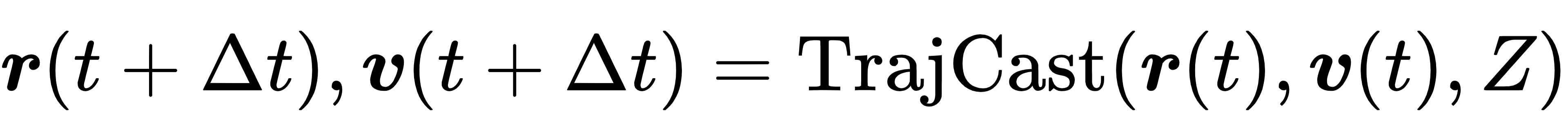
这就是无力分子动力学的核心公式——没有力、没有积分,直接一步推进原子轨迹。由于摆脱了数值积分的步长束缚,TrajCast可以使用5–30 fs的大步长,是传统MD的10–30倍。同样模拟1 ns轨迹,传统MD需要100万–200万步,而TrajCast仅需要3万–20万步,效率提升极为显著。
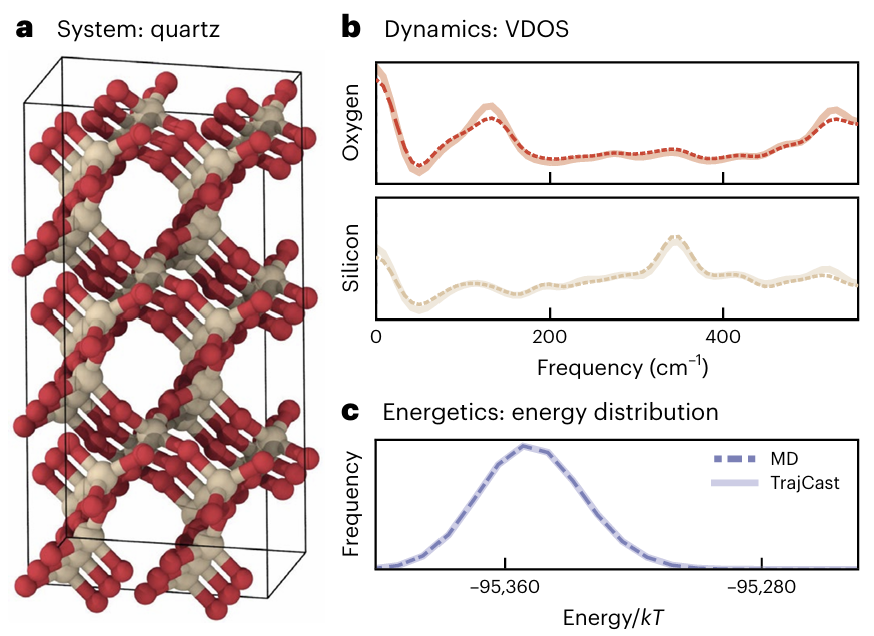
图2 TrajCast与传统MD的步长、效率对比图
2.5 隐性约束:玻尔兹曼分布
玻尔兹曼分布是统计力学的核心分布之一,描述了热力学平衡态下,系统中粒子处于不同能量状态的概率分布,其核心公式为:
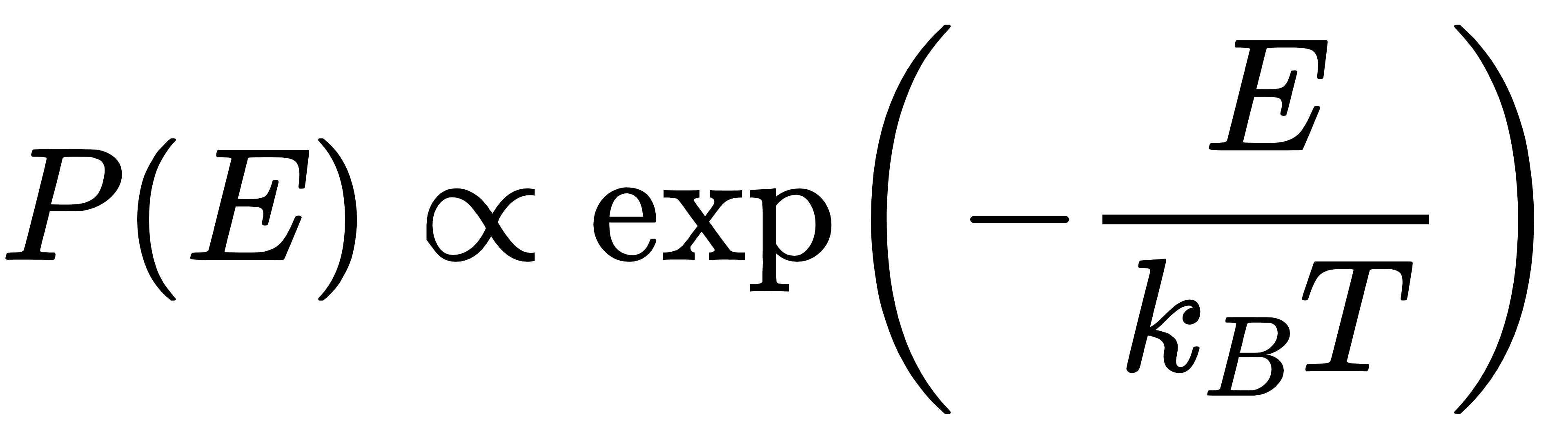
其中,P(E)是粒子处于能量为E状态的概率,kB是玻尔兹曼常数,T是热力学温度。该分布的核心意义的是:能量越低的状态,粒子出现的概率越高,这与原子系统的热力学特性高度契合。
TrajCast在模型训练和轨迹生成过程中,将玻尔兹曼分布作为隐性约束,确保生成的原子轨迹符合热力学平衡态特征。具体而言,模型会通过玻尔兹曼分布校正原子状态的概率分布,避免生成能量过高、不符合真实物理场景的原子构象,同时保证轨迹的能量守恒性和热力学合规性。这也是TrajCast能够精准复现液态水、晶体等体系热力学性质的关键原因之一——它不仅学习了原子的运动规律,更遵循了微观系统的热力学基本法则。
三、TrajCast全能性能验证:从小分子到玻璃态,零样本泛化突破极限
一款优秀的模拟工具,不仅要快,还要准。研究团队在三类完全不同的物理体系中,对TrajCast进行了严苛的benchmark测试,所有结果均与经典MD(参考标准)高度一致,充分证明了其高精度和广泛适用性。
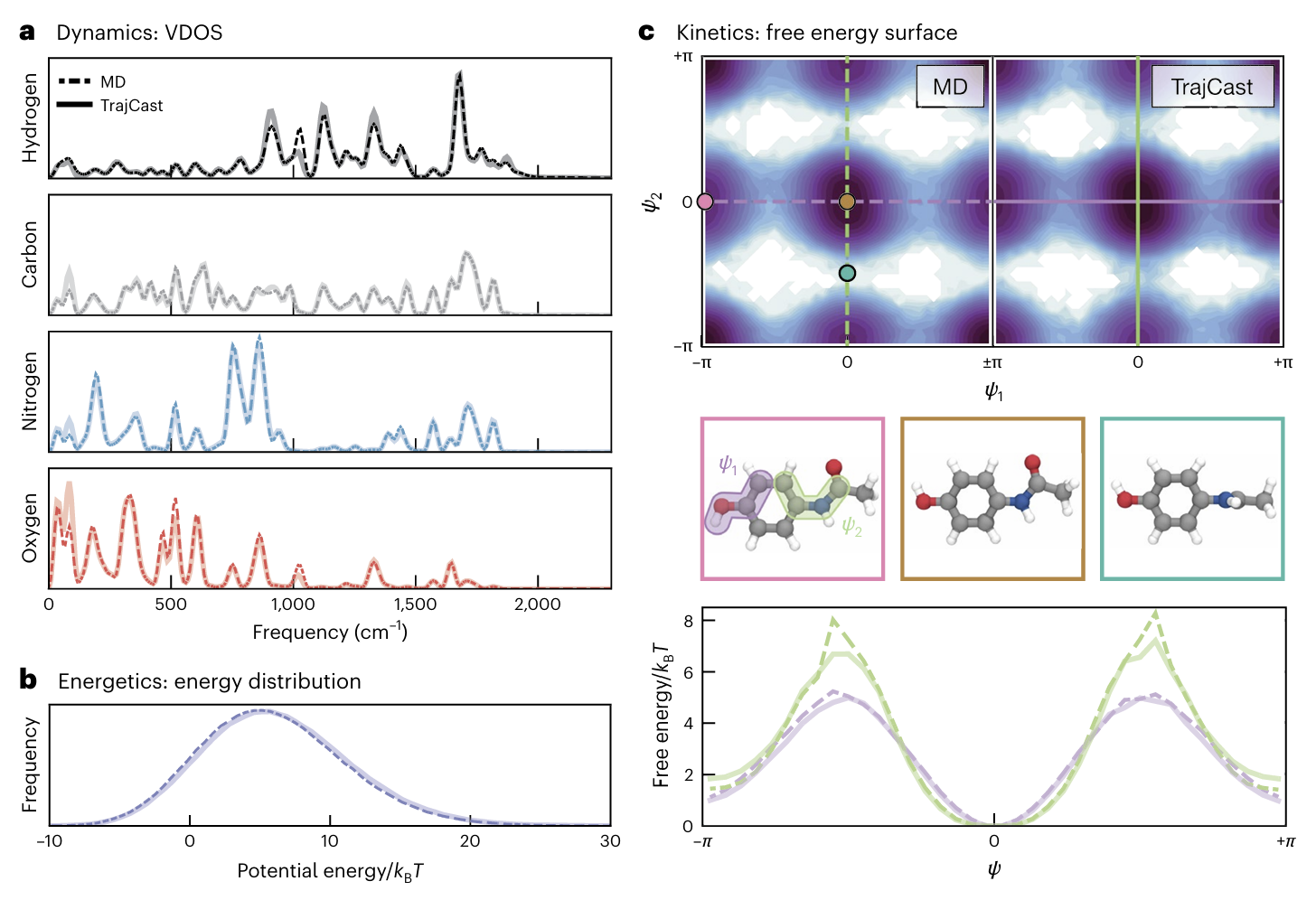
3.1 扑热息痛小分子:动力学、能量、自由能全对齐
扑热息痛是常见的小分子药物,其分子内存在胺基翻转等构象变化,对模拟精度要求较高。TrajCast使用7 fs的时间步长(是传统MD的14倍),稳定生成了7 ns的轨迹。测试结果显示:
1.振动态密度(VDOS)重叠度超过0.95,说明模型精准复现了分子的振动模式;
2.势能分布与经典MD几乎完全一致,能量守恒性良好;
3.二面角自由能垒误差小于kBT(k是玻尔兹曼常数,T是温度),处于热涨落范围内,完全符合物理现实;
最令人惊喜的是,训练数据中并不包含胺基翻转的高能构象,但TrajCast在零样本条件下,自动生成了正确的翻转结构,证明其具备极强的外推能力,不是简单拟合训练数据,而是真正学习到了分子运动的底层规律。
3.2 α-石英晶体:30 fs大步长,大体系高效模拟
α-石英是典型的共价晶体,原子排列高度有序,运动受限较强,对模拟的稳定性要求极高。TrajCast将时间步长进一步提升至30 fs(是传统MD的30倍),在长达15 ns的轨迹中保持高度稳定。测试结果显示:
1.振动谱重叠度超过0.95,与经典MD几乎重合;
2.能量分布始终稳定,没有出现明显漂移;
3.对于包含4300原子的超大超胞,TrajCast单日可生成15 ns以上的轨迹,远超当前主流机器学习势函数(如NequIP、Allegro)的模拟效率,解决了大体系长时程模拟的痛点。
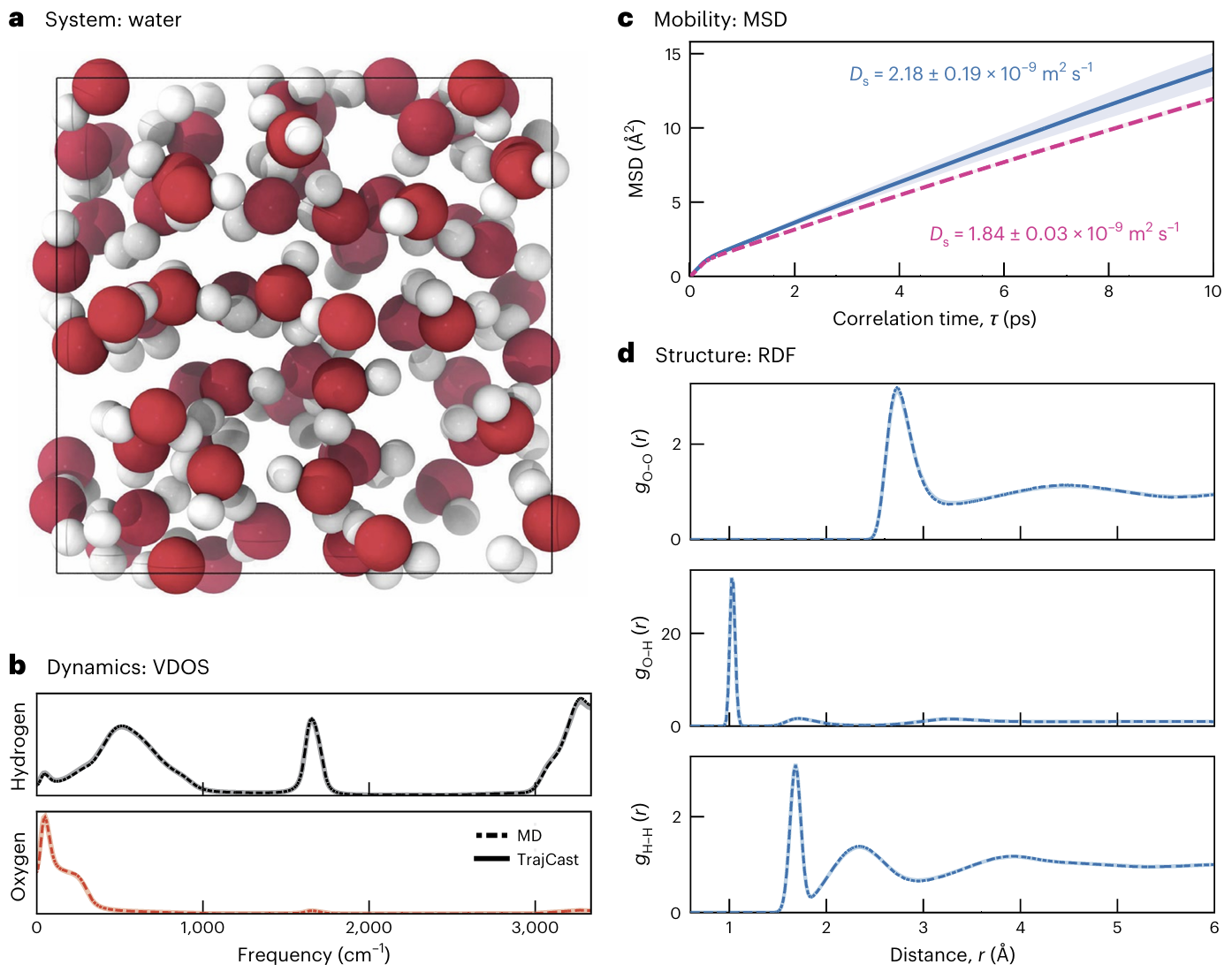
3.3 液态水:结构、扩散、径向分布函数精准复现
液态水是最常见也最复杂的分子体系之一,其分子间存在氢键作用,动力学行为复杂,对模拟精度的要求极高。TrajCast使用5 fs的时间步长(是传统MD的10倍),测试结果显示:
1.扩散系数为2.18×10⁻⁹ m²/s,与经典MD(2.20×10⁻⁹ m²/s)几乎一致;
2.O–O、O–H、H–H三种径向分布函数(RDF)与经典MD高度重合,精准复现了水分子的局部结构;
3.振动谱重合度约为0.99,完美捕捉了水分子的振动特征。
3.4 零样本封神:仅训常温液态水,复现超淬冷水玻璃化
这是TrajCast最具突破性的表现。玻璃化转变是材料科学中的经典难题,指液体快速降温时,分子排列从无序液态转变为无序固态(玻璃态)的过程,需要模拟跨温区的慢动力学行为,传统模拟工具往往需要大量低温数据训练才能实现。
而TrajCast仅使用300 K(常温)液态水的数据进行训练,没有加入任何低温、玻璃态数据,直接将模型用于快速降温模拟(从300 K降至180 K),成功复现了水的玻璃化转变过程:
1.扩散系数随温度的变化曲线与经典MD完全一致,低温下扩散系数急剧下降,符合玻璃化转变的特征;
2.O–O径向分布函数的演化的与经典MD高度吻合,从液态的短程有序、长程无序,逐渐转变为玻璃态的短程有序、长程更无序;
3.结构序参数的拓扑路径与经典MD一致,证明模型准确捕捉了玻璃化转变的核心物理机制。
这一结果充分证明,TrajCast真正学习到了原子运动的底层物理规律,而非简单拟合训练数据,其零样本泛化能力远超传统机器学习势函数。
四、范式革新:无力MD开启原子模拟第三代路线
纵观原子模拟的发展历程,已经经历了两代技术范式,而TrajCast的出现,标志着第三代范式的诞生,三者的对比清晰可见:
第一代:经典力场MD。核心是基于经验势函数计算力,速度快但精度有限,难以描述化学反应、氢键等复杂相互作用,适用于简单体系的快速模拟。
第二代:机器学习势函数MD(MLIP-MD)。核心是用AI模型拟合第一性原理数据,精度接近第一性原理,但仍然需要计算力、进行数值积分,步长受限,无法突破效率天花板,适用于中等精度、中等体系的模拟。
第三代:无力AI动力学(TrajCast)。核心是直接预测原子轨迹,不算力、不积分、大步长,同时实现高精度、强泛化,适用于大体系、长时程、非平衡态等传统MD难以覆盖的场景。
TrajCast的范式革新,其科学价值远不止“更快”,更在于它重新定义了原子模拟的底层逻辑:从“基于势能面求导”转向“基于相空间演化学习”,让模拟不再依赖算力堆砌,而是依靠AI对物理规律的学习,大幅降低了长时程、大体系模拟的门槛。
这种革新将深刻影响多个领域:在材料科学中,可高效模拟玻璃化、相变、离子输运等过程,加速新型材料开发;在生物医药中,可快速模拟蛋白质折叠、配体结合等过程,助力新药研发;在催化领域,可精准预测催化剂活性位点与反应路径,提升催化效率。
五、量子赋能:进一步突破经典模拟极限
TrajCast已经实现了经典AI驱动下的原子模拟革命,而借助量子计算相关技术,可进一步突破经典AI的性能上限,实现“量子+无力MD”的融合创新,让模拟效率、泛化能力再上一个台阶。其中,玻色量子的量子采样能力与量子+AI融合功能,是赋能TrajCast的核心关键。

图5 新一代专用量子计算机“驭量·山海1000”发布现场
玻色量子是国内领先的光量子计算机企业,其核心产品“驭量·山海1000”是国内首个千比特级可扩展专用量子计算机,采用室温全光纤架构,具备连续变量量子态、量子机器学习等核心能力,特别适合分子模拟、材料发现等场景。
玻色量子的量子采样能够在毫秒级遍历经典计算机难以抵达的高维构象空间。与传统经典采样相比,它无需遍历所有可能的原子构象,可直接高效筛选出能量较低、符合物理规律的优势构象,采样效率提升数个数量级——这对于原子模拟中“构象搜索”这一核心痛点,是极具针对性的解决方案。
此外,玻色量子将量子计算与AI模型深度融合,打造了量子机器学习平台,可实现“量子采样+经典AI”的协同优化:一方面,用量子采样为实验提供高质量的构象先验,解决经典AI采样效率低、易陷入局部最优的问题;另一方面,用经典AI对量子采样得到的构象进行动力学推进,弥补量子计算在长时程演化模拟中的不足,形成“量子赋能AI、AI落地量子”的闭环。
具体量子计算到对TrajCast的赋能,主要体现在两个核心方面:一是加速TrajCast的等变图神经网络推理,快速提取原子局部环境的关键信息,让模拟速度再提升一个数量级;二是生成经典难以高效获取的低温、相变等构象先验,注入TrajCast模型,进一步提升其零样本泛化能力,让模型在极端场景中依然保持高精度。
这种融合无需对TrajCast进行大规模改造,可快速落地,形成“量子采样+AI动力学”的混合模拟引擎,进一步压缩模拟时间,推动原子模拟从“AI加速”迈向量子增强时代,为高端材料、新药研发等领域提供更高效的工具。
六、总结
TrajCast提出的无力分子动力学框架,是原子模拟领域具有里程碑意义的范式突破。它抛弃了传统MD必需的力计算与数值积分,通过自回归等变神经网络直接预测原子运动,嵌入E(3)等变、速度显式建模、守恒律约束三大核心设计,同时结合玻尔兹曼分布保证热力学合规性,实现了“不算力、不积分、大步长、高精度、强泛化”的统一。
从扑热息痛小分子到α-石英晶体,从液态水到超淬冷水玻璃化,TrajCast在多个体系中展现出超越传统方法的性能,重新定义了原子模拟的效率与精度上限。而量子计算技术凭借其高效的量子采样能力与量子+AI融合功能,为TrajCast提供了经典AI无法实现的增强路径,进一步突破模拟极限。
未来,原子尺度模拟将不再依赖传统算力堆砌,而是由AI与量子计算协同驱动,以极低的成本、极高的效率探索微观世界。这一变革不仅会重塑材料科学、化学、生物医药的研究范式,更将加速关键核心材料与药物的发现进程,为科学与产业带来真正的跨越式发展。
论文链接:https://www.nature.com/articles/s42256-026-01227-7 | 





















