|

原文地址:https://doi.org/10.1038/s41524-026-02131-9
代码地址:https://github.com/JieCoa/AtomNet
研究背景与科学问题
晶体材料性质预测是加速新材料发现的核心环节。传统基于密度泛函理论(DFT)的第一性原理计算虽精度高,但其高昂的计算开销严重限制了对候选材料的大规模筛选。近年来,图神经网络(GNN)及其Transformer变体在材料结构研究中表现出卓越性能,成为高通量材料计算的重要工具。
然而,现有方法面临一个关键瓶颈:如何系统地将晶体性质相关的物理化学属性融入图模型,并在保持模型通用性的同时避免冗余。一方面,现有研究多采用原子序数或CGCNN中的原子嵌入对节点初始化,忽视了不同物理属性对不同晶体性质的差异化贡献;另一方面,传统机器学习的特征重要性分析方法难以揭示GNN内部学习到的复杂依赖关系。针对这些挑战,集美大学黄楷团队提出AtomNet——一个物理信息驱动的多边图神经网络框架,通过协同优化特征设计、晶体图构建与模型架构,显著提升了晶体性质预测的精度与可解释性。
AtomNet整体架构与设计思路
AtomNet的整体架构如Fig.1所示
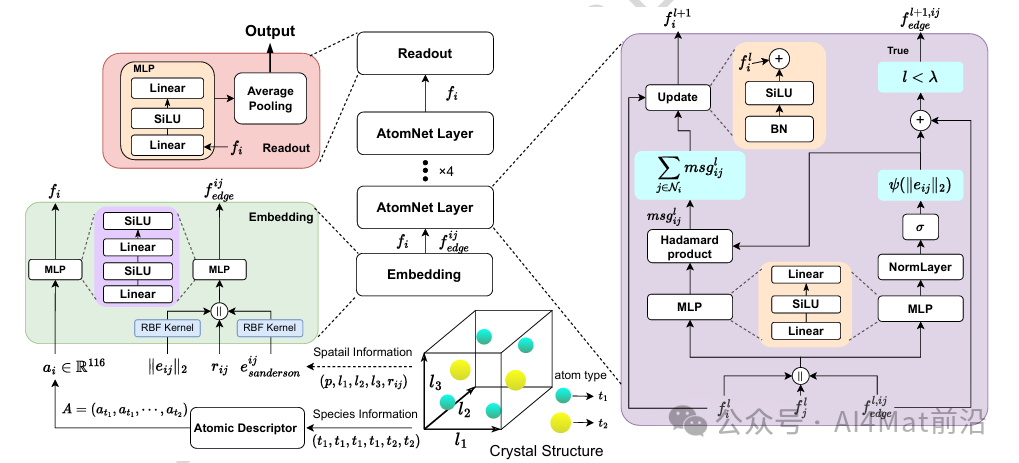
▲ Fig.1 | AtomNet架构示意图。在晶体结构中,t1和t2表示不同的原子类型,p表示原子的三维坐标,l1、l2、l3对应晶格L的三个晶格矢量,rij表示原子i和j之间的边矢量。其中j ∈ Ni,Ni为中心原子i的邻域。原子描述符对应于方法部分中的物理化学原子描述符,输出由原子类型t1、t2推导出的116维原子描述符。原子
该框架以晶体结构为输入,通过原子描述符模块将原子类型转化为116维的物理化学属性向量,作为图节点的初始特征;同时基于晶格矢量与原子坐标构建多边晶体图,引入原子电负性差异作为新颖的边特征。模型通过消息传递机制聚合邻域信息,最终输出对目标性质的预测。
与现有工作相比,AtomNet的核心创新体现在三个层面:节点表征层面,通过精心设计的物理化学属性组合显式编码原子内禀特性;边表征层面,引入电负性差异捕捉成键极性信息,且不显著增加参数规模;图构建层面,采用确定性周期模式方案与距离衰减权重函数,确保邻域结构的稳定性并抑制远端噪声。
物理化学原子描述符的构建
AtomNet在节点初始化阶段摆脱了对单一原子嵌入的依赖,转而采用包含基本物理与化学属性的116维原子描述符。这些属性涵盖了原子半径、电离能、电负性、电子亲和能、价电子排布等多种与晶体性质密切相关的物理量。这种设计的科学动机在于:晶体性质源自所有原子属性的协同效应,且不同性质对应的关键属性各异,仅依赖单一属性难以准确表征多元化的目标。
为了量化各属性对预测的实际贡献,作者采用基于梯度的积分梯度(Integrated Gradients, IG)方法进行可解释性分析。该方法通过沿基线输入到实际输入的线性插值路径计算梯度积分(如Fig 2所示),获得每个属性对模型预测结果的归因强度。相比传统基于相关性的特征重要性方法,IG能够揭示GNN内部学习到的非线性依赖关系,从物理层面验证模型决策的合理性。
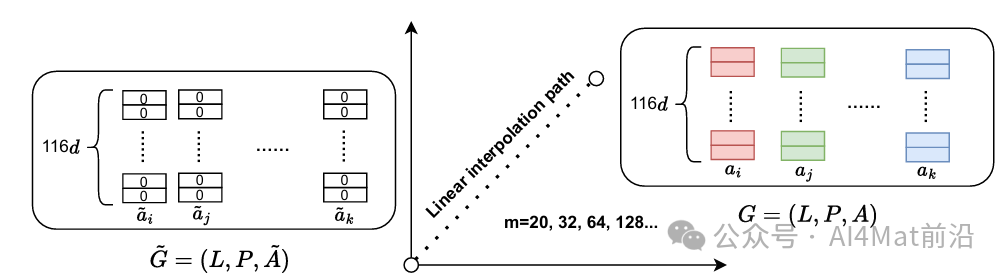
▲ Fig.2 | 从基准输入˜G到实际输入G的线性插值路径。基准输入G˜中所有节点的特征均设置为0,而实际输入G对应于所有节点的特征值(独热编码)。m为积分步数,对应于插值点的数量。A˜ = [˜ai, ˜aj, · · · , ˜ak]T表示基准输入。
多边晶体图构建与边权重函数
晶体图的构建是AtomNet另一核心环节。如Fig.3 所示,作者基于截断半径策略定义中心原子的邻域,将周期性晶格矢量纳入图结构表示。然而,固定截断半径在多边图中可能导致非唯一的邻域形成。为此,AtomNet引入确定性周期模式方案,确保每个中心原子的邻域结构稳定唯一。
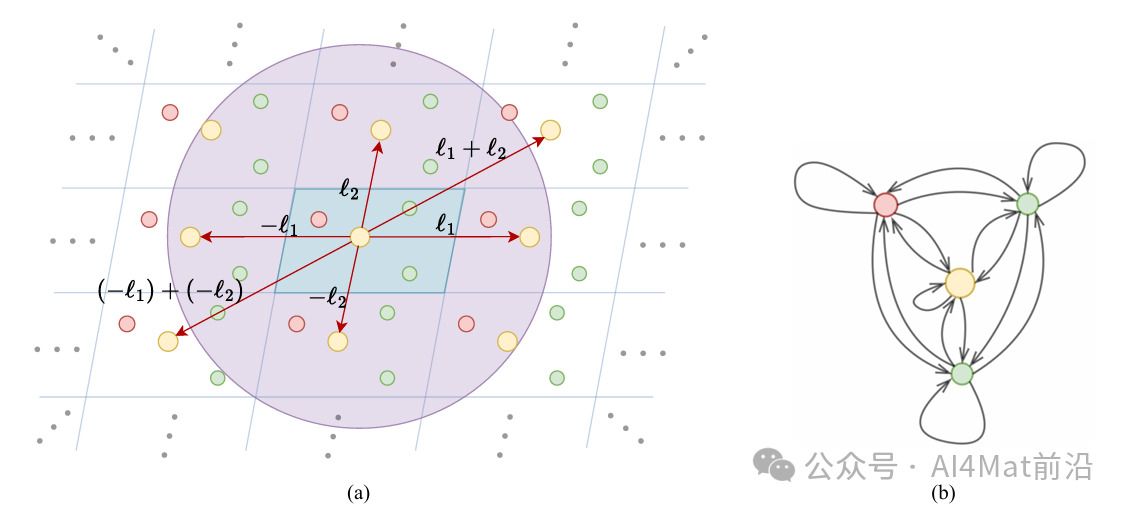
▲ Fig.3 | 晶体图的示意图。红色箭头表示二维晶体结构中的周期性模式ℓ1和ℓ2。蓝色四边形对应于原始晶胞的二维表示,并从蓝色四边形周期性延伸。不同颜色的小圆圈代表不同的原子类型,紫色圆形区域表示以黄色圆圈为中心原子的截断半径邻域内的范围。(a) 具有半径邻域的晶体结构
为了进一步分析边长分布对模型的影响,作者统计了Jarvis数据集中晶体图的边长分布(Fig. 4)。结果表明,截断半径附近存在大量边,这些远端邻居可能引入噪声并削弱核心原子间相互作用的表达能力。
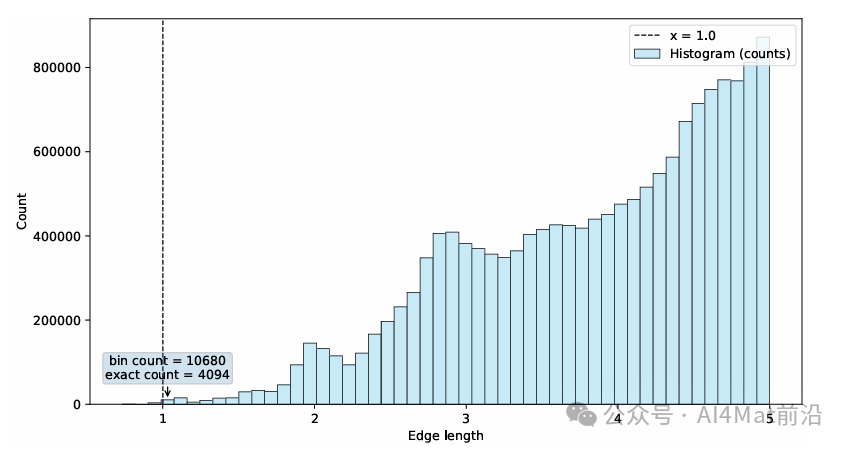
▲ Fig.4 | 在Jarvis数据集(形成能任务,训练集包含44,578个晶体数据)上构建的晶体图中,截断半径内边长分布的直方图。x = 1.0处的虚线对应于公式(20)中的分段点d0。图中方框标注报告了两个统计量:落在包含x = 1.0的直方图区间内的条目数(区间计数)以及x ∈ [0.98, 1.02]范围内的条目数(精确计数)。
针对这一问题,AtomNet提出距离衰减边权重函数,包括Simply与Cubic Smooth两种实现形式(Fig. 5)。该函数通过赋予近距离原子对更高权重、远距离原子对更低权重,实现了对核心原子相互作用的优先建模。Cubic Smooth函数在截断半径附近平滑衰减,有效抑制了边界噪声;同时,权重函数的引入也提升了训练效率。
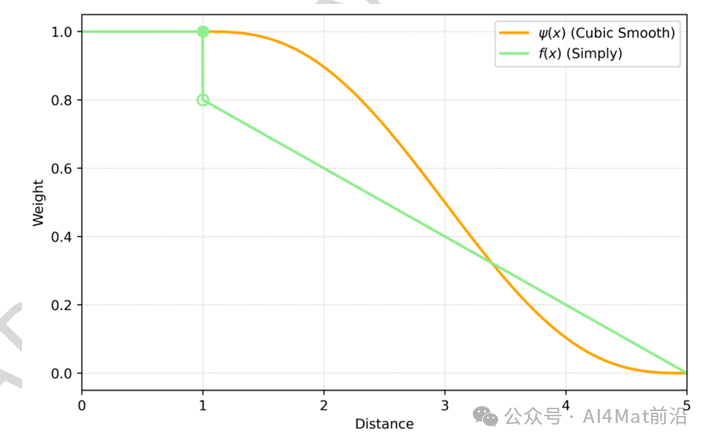
▲ Fig.5 | 在截断半径rcut = 5 Å内,Simply函数和Cubic Smooth函数边权重变化曲线。绿色实心圆点表示Simply函数在x = 1.0处(定义点)的左极限值为1.0。绿色空心圆点表示Simply函数在x = 1.0处的右极限值为0.8。
值得一提的是,AtomNet在边特征中引入了电负性差异这一物理量。电负性差反映了原子间成键的极性强度,是预测电子性质的重要物理先验。相较于引入高阶几何特征(如键角、二面角等),电负性边特征在不显著增加参数量与训练开销的前提下,显著增强了模型对晶体内化学键合特征的捕捉能力。
性能评估与对比实验
作者在两个标准材料数据集——Jarvis和Materials Project (MP)上对AtomNet进行了全面评估,对比基线包括Matformer、PotNet、ComFormer、Crystalformer、CrystalFramer与CartNet等当前领先方法。
▲ Table 1 | Jarvis数据集上的性质预测结果。
如Jarvis数据集结果所示,AtomNet在五项任务(Formation Energy、Total Energy、Bandgap (OPT)、Bandgap (MBJ)、Ehull)上均取得最佳表现。其中Ehull任务的提升最为显著,相比次优模型CartNet降低误差达20.3%;在低数据量的Bandgap (MBJ)任务中,AtomNet亦实现2.9%的性能提升;Formation Energy、Total Energy与Bandgap (OPT)任务分别实现5.0%、3.4%与7.0%的MAE降低。
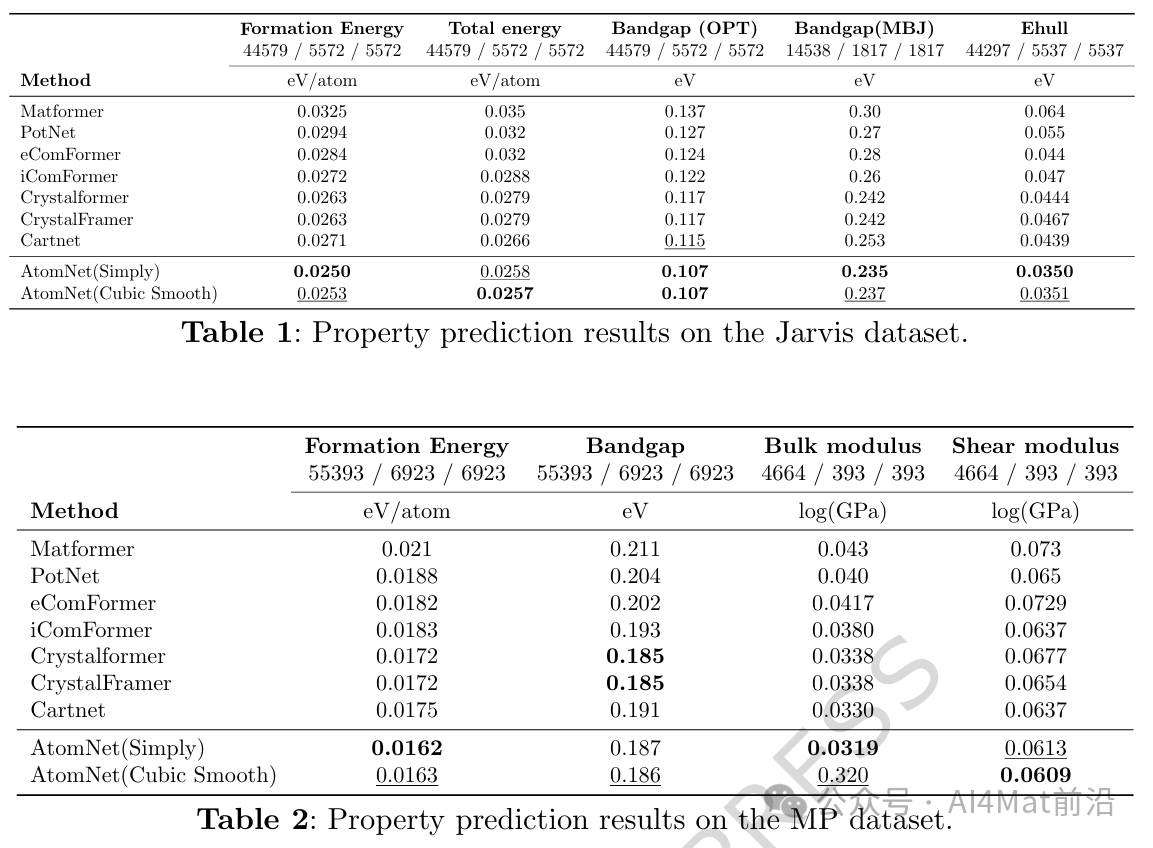
在MP数据集上,AtomNet除Bandgap任务外的所有任务均表现最优。Formation Energy任务上的MAE降低约5.8%;在仅有4,664训练样本的Bulk modulus与Shear modulus任务中,分别实现3.3%与4.4%的误差降低,体现了AtomNet在小样本场景下的良好泛化能力。这一结果验证了富含物理属性的原子描述符与边权重函数能够有效提升模型在数据有限条件下的学习能力。
计算效率与消融分析
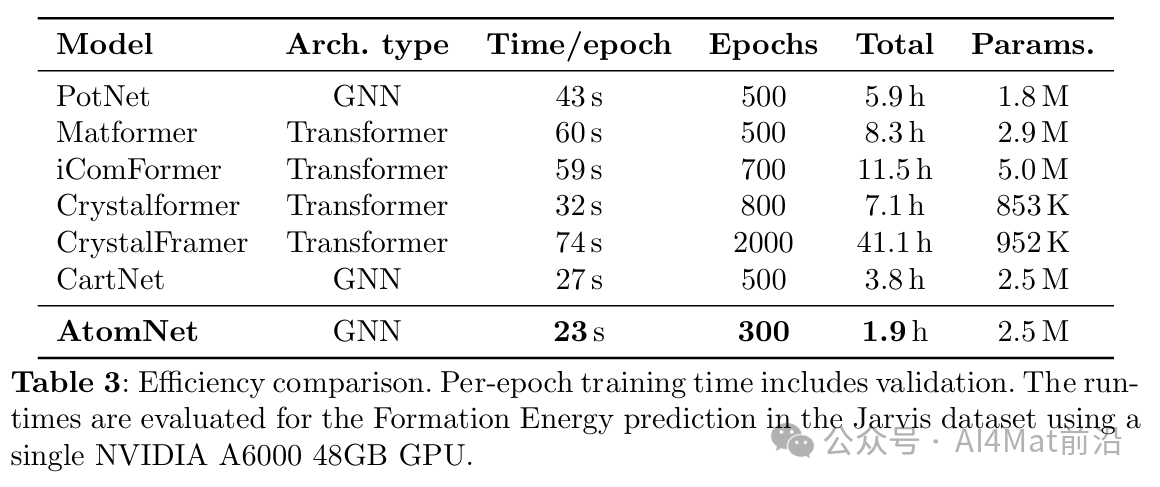
效率对比实验显示,AtomNet在保持高精度的同时具有合理的计算开销。相比之下,CrystalFramer等基于Transformer架构并集成角度信息的方法虽然精度接近,但训练资源需求显著更高。
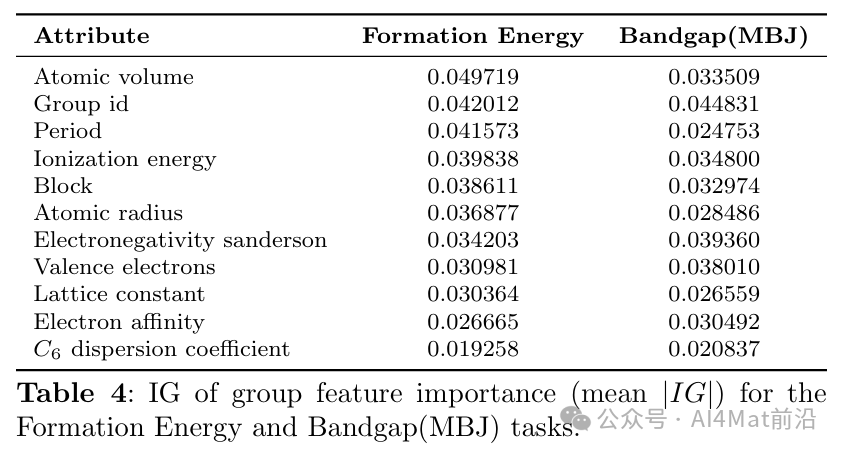
为深入剖析各组件的贡献,作者开展了系统的消融研究。通过IG方法计算各属性组的特征重要性(Table 4)以及属性剪枝实验(Table 6),结果证实了原子描述符中各物理化学属性的差异化贡献。
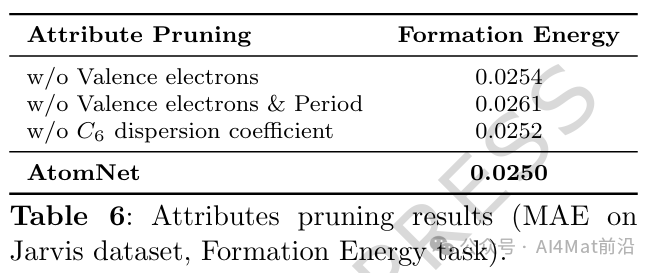
消融实验进一步验证了原子描述符、边权重函数与电负性边特征三者均为模型性能提升的关键因素。其中,移除任一模块均会导致预测精度的明显下降,说明这些设计在捕捉晶体物理规律方面具有协同效应。
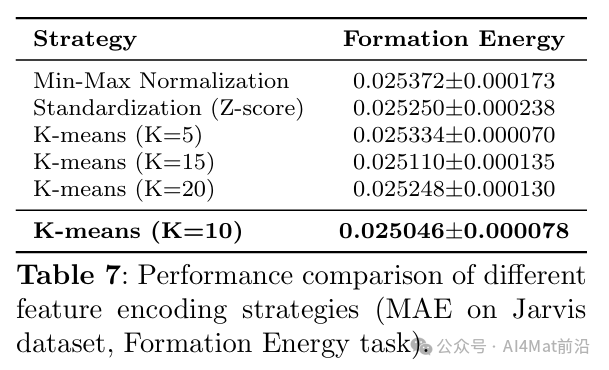
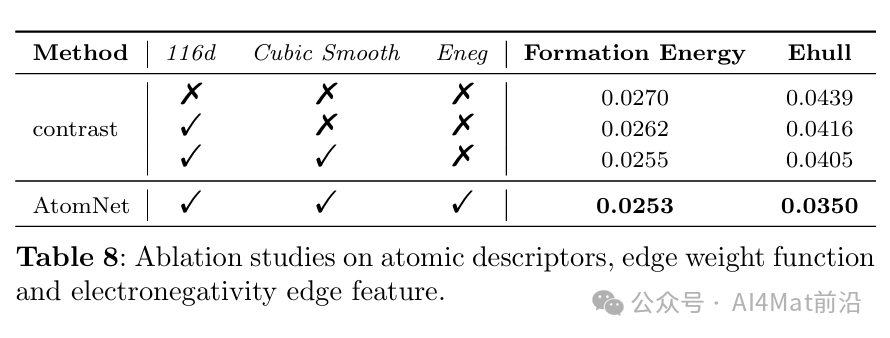
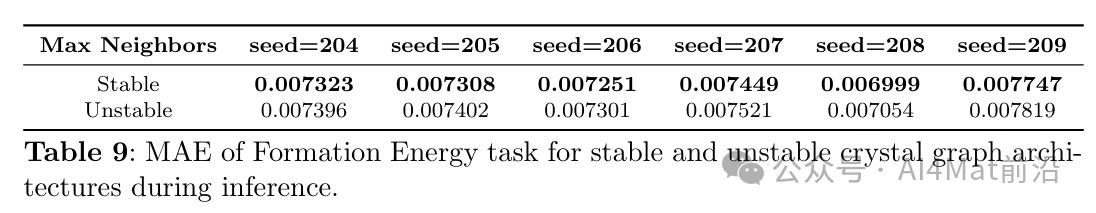
针对晶体图构建中的稳定性问题,作者还比较了稳定与非稳定晶体图架构在Formation Energy任务上的表现(Table 9),结果表明确定性周期模式方案显著改善了模型的鲁棒性。
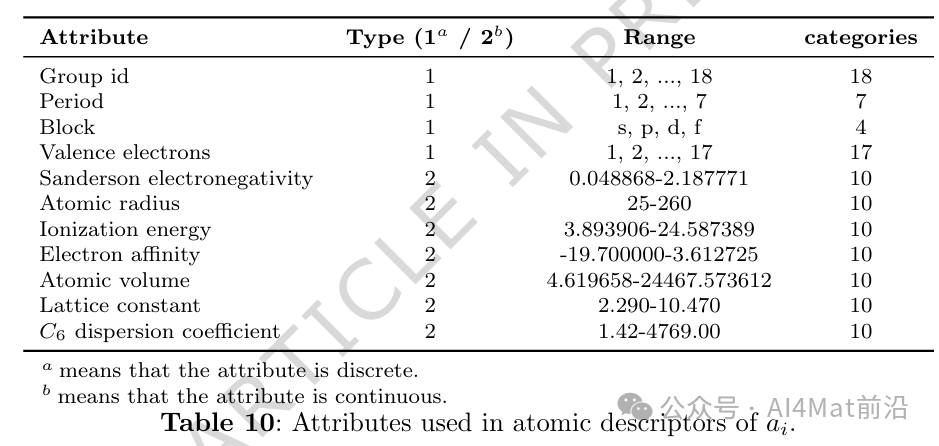
详细列举了原子描述符中所采用的具体物理化学属性,为后续研究者复现与扩展提供了清晰参考。
研究意义与展望
本研究的核心贡献可归纳为三个方面:
第一,方法学创新。 AtomNet首次系统地将物理化学属性整合至图神经网络的节点表征中,构建了116维富含物理意义的原子描述符;同时引入电负性差异作为边特征,从化学键合层面增强了模型表征能力。 第二,可解释性提升。 通过积分梯度方法,作者建立了"原子物理属性—模型决策—宏观晶体性质"的因果链条,揭示了不同物理属性对不同晶体性质预测的贡献规律,提升了机器学习模型在材料科学中的物理可信度。 第三,工程实践价值。 AtomNet在Jarvis与MP两大基准上均取得领先性能,尤其在Ehull、Formation Energy等关键任务上实现显著提升,且在小样本任务中展现出良好的泛化能力,为加速新材料发现提供了有效工具。 研究局限与未来方向: 尽管AtomNet在多任务上表现出色,但在Bandgap等部分任务上与基于Transformer的最优模型存在微小差距,提示模型在长程相互作用建模方面仍有改进空间。未来研究可考虑:(1) 将原子描述符扩展至更复杂的多体物理量(如自旋-轨道耦合相关属性);(2) 探索物理信息先验与自注意力机制的深度融合;(3) 将该框架推广至缺陷晶体、无序固溶体等更复杂的材料体系。
对相关研究者而言,本文的启示在于:物理先验与数据驱动学习的协同优化是材料人工智能领域的重要范式。在追求模型架构创新的同时,深入挖掘领域知识、构建可解释的特征工程方案,将是推动该领域持续发展的关键路径。
参考文献:Cao, J., Huang, K., Mao, J. et al. Physics-informed graph neural network representation learning for crystal property prediction.npj Comput Mater*(2026). https://doi.org/10.1038/s41524-026-02131-9
文章改编转载自微信公众号:AI4Mat前沿
原文链接:https://mp.weixin.qq.com/s/G0yNnP-2t4ZXvFENMWChng?scene=1&click_id=27 | 





















