本文解读研究《 Coarse-Grained Boltzmann Generators 》。该模型统一了粗粒度降维与玻尔兹曼生成的双重优势,在低维粗粒空间中完成高效生成式采样,并通过学习得到的平均力势(PMF)进行严格重加权,最终实现可扩展、高效率、热力学无偏的分子平衡态采样。CG-BG 可以直接从加速模拟数据训练,不需要漫长的全原子平衡模拟,同时能精准捕获溶剂介导效应等复杂多体相互作用,在 Müller–Brown 势场与丙氨酸二肽显式溶剂体系中全面超越传统隐式溶剂模型,为蛋白质构象系综、高分子动力学、生物分子相互作用等大分子模拟难题,提供了一条 “又快又准” 的全新路径。

在计算生物物理、药物设计与材料科学中,最核心、最困难的任务之一,就是正确、高效地从玻尔兹曼分布中采样分子平衡构象。无论是蛋白质如何折叠、药物分子如何与靶点结合、溶液环境如何改变分子构象,都必须建立在正确的平衡分布之上。然而,真实分子体系自由度极高、势能面极其崎岖,传统分子动力学(MD)往往要花费数周甚至数月才能勉强收敛;而近些年出现的玻尔兹曼生成器虽然精度极高,却受限于维度灾难,几乎无法扩展到大分子;粗粒度模型速度极快,却因为缺少严格的重加权步骤,结果永远带有统计偏差。
长期以来,整个领域都被困在一个无法调和的矛盾里:全原子模型准到可信,但慢到无法使用;粗粒度模型快到实用,但偏差大到不可靠。
而 CG-BG(粗粒度玻尔兹曼生成器) 第一次真正打破了这一困局。它把粗粒度的速度、玻尔兹曼生成的精度、增强采样的高效性,统一在一套严格的统计物理框架中,让 “又快、又大、又准” 的分子模拟第一次成为现实。
一、背景:分子平衡采样的三大核心基础
要理解 CG-BG 的革命性,我们必须先看清分子模拟最底层的三个关键概念:玻尔兹曼分布、玻尔兹曼生成器、粗粒度边际分布。
1.1 玻尔兹曼分布:所有平衡态的唯一真理
任何热力学平衡的分子系统,构象出现的概率都必须服从玻尔兹曼分布:
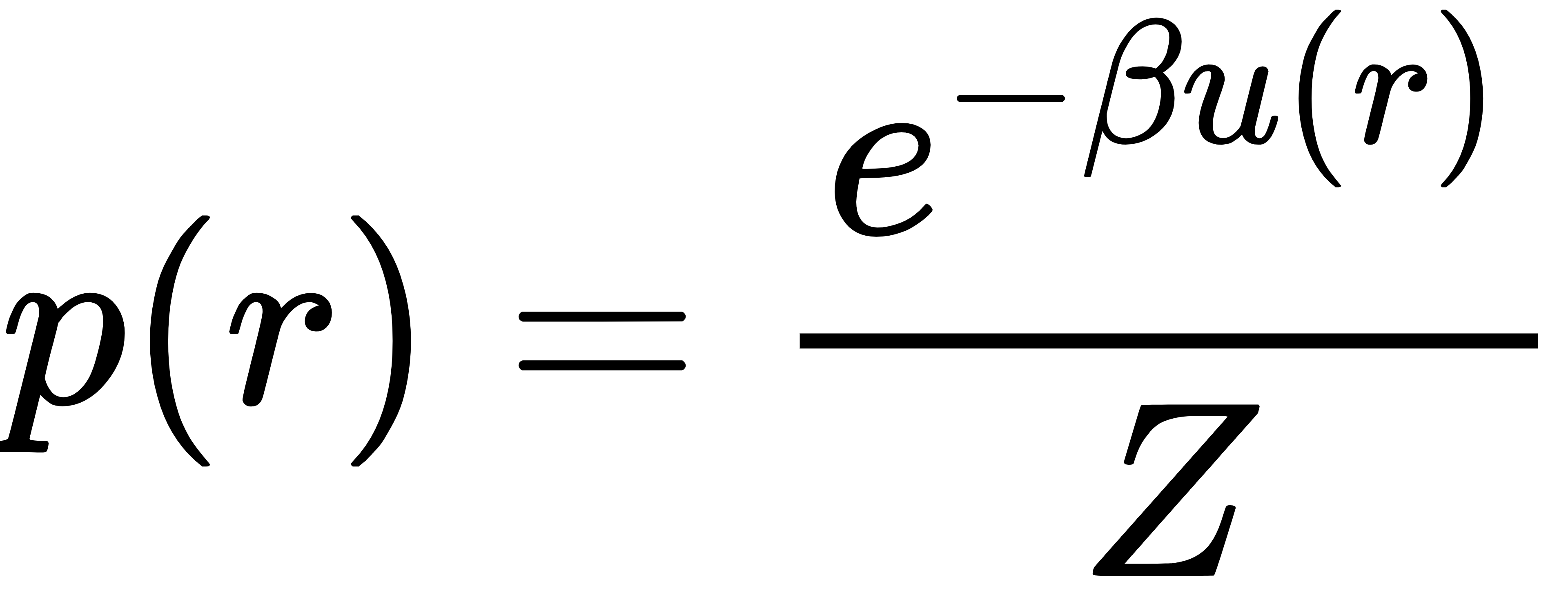
其中 r 是原子坐标,u(r)是势能,β=1/(kBT)由温度决定,Z 是配分函数。这个公式告诉我们:能量越低的结构,出现概率越高。所有模拟、计算、生成模型的终极目标,都是尽可能接近这个分布。但因为体系维度极高,直接计算几乎不可能。
1.2 玻尔兹曼生成器:用 AI 直接生成平衡态
玻尔兹曼生成器(BG)使用归一化流模型,直接学习从简单分布(如高斯)映射到分子构象,然后用重要性采样对生成样本加权,从而恢复真实的玻尔兹曼分布。它的核心权重公式为:
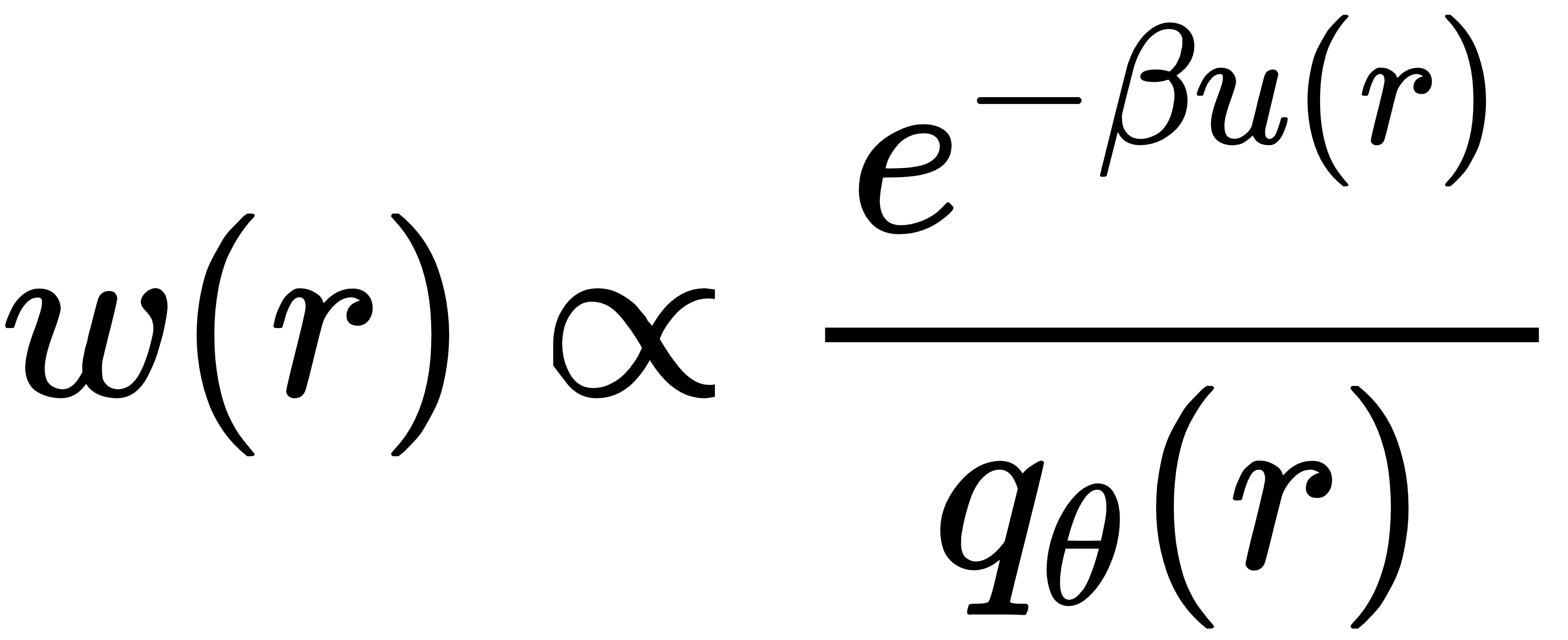
其中 qθ 是 AI 生成的分布。通过加权,模型可以获得完全无偏的热力学结果。但它的致命弱点是:体系一大、原子一多,计算量立刻爆炸,无法扩展。
1.3 粗粒度建模:把大分子 “降维”
粗粒度(CG)将一堆原子映射成少数 “珠子”,把高维原子坐标 r 投影到低维粗粒坐标 R:
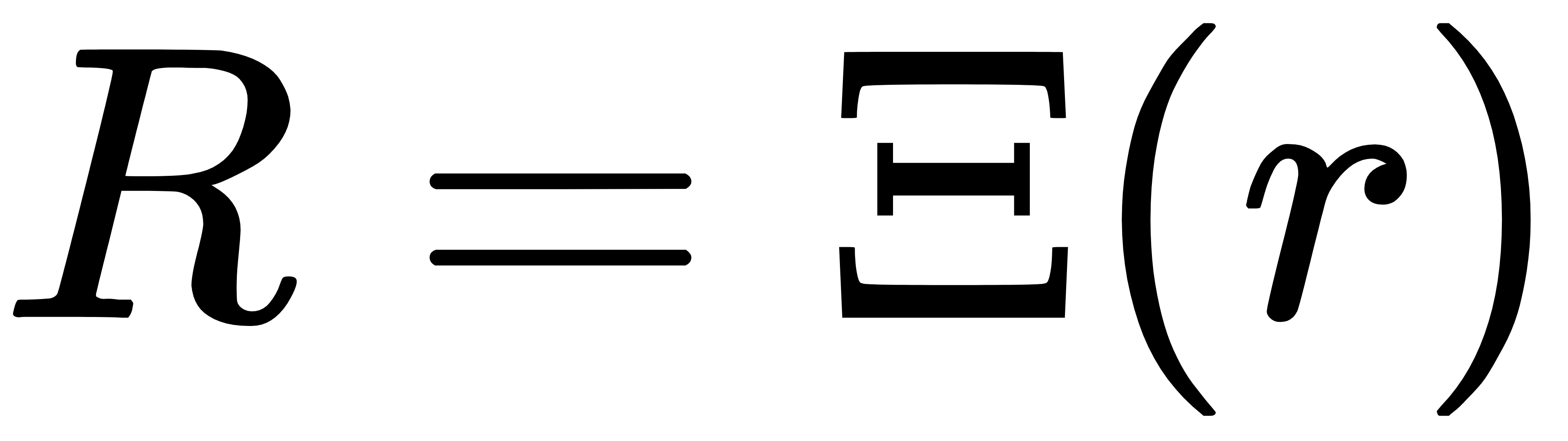
维度下降后,速度可以提升几十倍。但传统粗粒度模型最大的问题是:没有正确的重加权机制,无法还原真实平衡分布,因此结果永远存在偏差。
二、CG-BG 核心创新:统一粗粒度与玻尔兹曼生成
CG-BG 的核心思想极其简洁优雅:在粗粒度空间做玻尔兹曼生成,用学习到的平均力势做严格重加权。它第一次把 “降维提速” 和 “热力学无偏” 合二为一。
2.1 整体架构:三大模块构成完整系统
CG-BG 由三个协同工作的模块组成:
粗粒度映射:把原子坐标打包成低维粗粒珠子;
PMF 网络:学习粗粒空间的 “有效能量”—— 平均力势;
流生成模型:在粗粒空间快速生成构象。
最终,生成的构象通过重加权校正,直接输出无偏、稳定、热力学严格的平衡分布。
2.2 关键创新 1:用力匹配学习平均力势(PMF)
粗粒空间的目标能量不是普通势能,而是平均力势(PMF),它包含被消除掉的原子带来的熵与能量贡献。CG-BG 使用变分力匹配训练 PMF 网络:
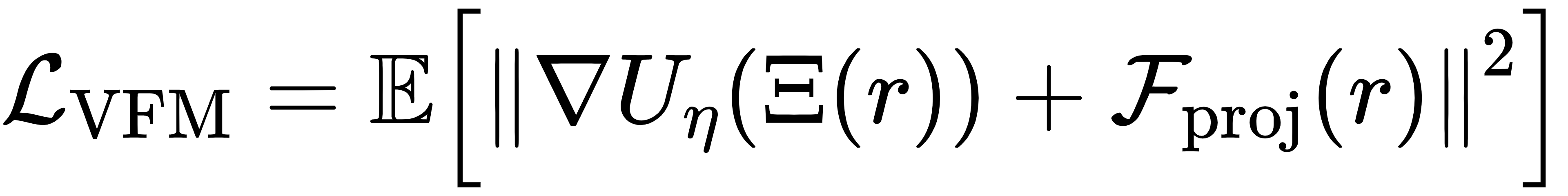
这个公式的含义是:让粗粒空间的力,尽可能逼近真实原子力在粗粒空间的平均投影。更重要的是,即使数据来自加速模拟(偏置数据),仍然可以学到无偏 PMF,这让训练成本大幅下降。
2.3 关键创新 2:粗粒度空间重加权,实现无偏采样
CG-BG 继承玻尔兹曼生成器的精髓,在粗粒空间进行严格重加权:
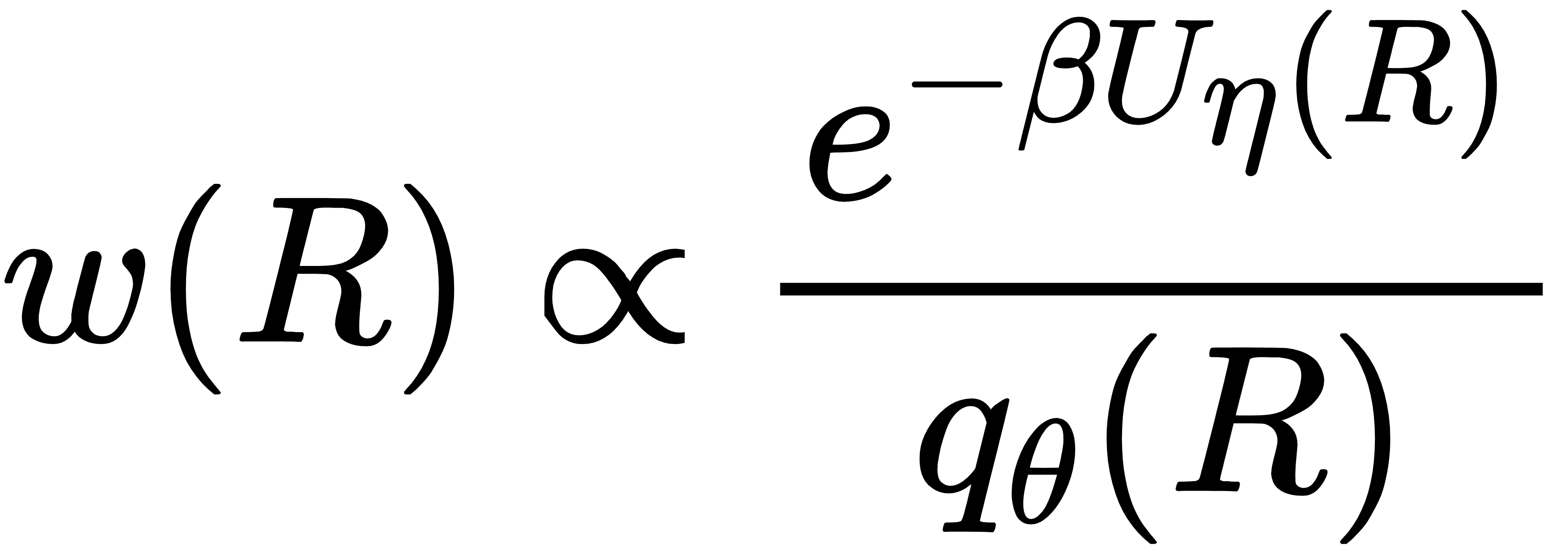
其中 Uη(R)是学习到的 PMF,qθ(R)是流模型生成的分布。只要 PMF 足够准确,加权后的分布就会严格收敛到真实的物理平衡分布。这是 CG-BG 能够 “又快又准” 的核心原因。
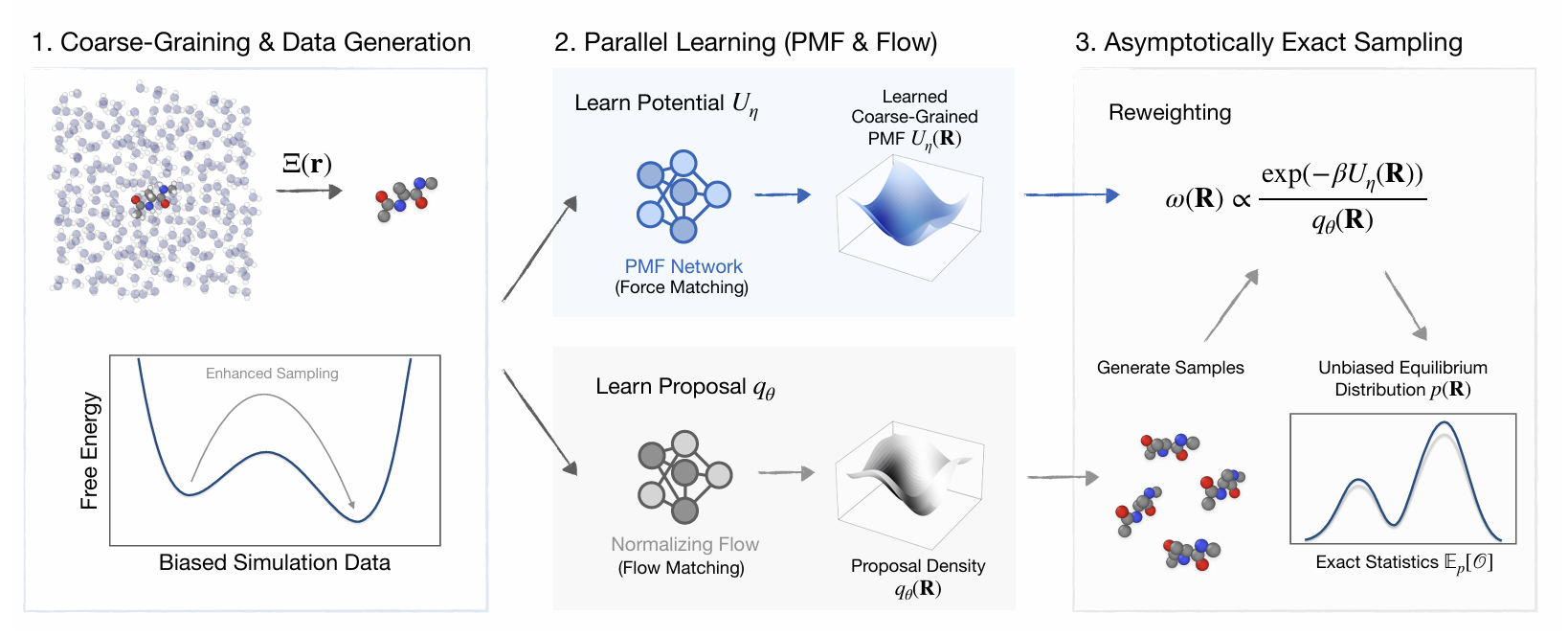
图1 CG-BG整体工作流程
三、CG-BG 完整算法流程:训练与采样
CG-BG 的运行流程清晰、可复现、可扩展,分为三个阶段。
3.1 数据准备与粗粒映射
首先从增强采样模拟中快速收集构象数据,不需要长时间的平衡模拟。随后将原子坐标映射为粗粒坐标,把高维体系转为低维体系,大幅降低后续学习难度。同时计算原子力在粗粒空间的投影,用于后续训练。
3.2 并行训练:PMF 网络与流模型一起学习
在训练阶段,CG-BG 同时训练两个模型:
一是 PMF 网络,用力匹配损失学习粗粒空间的有效能量;
二是流生成模型,用流匹配学习构象分布。
两个模型互不干扰、可以并行训练,大幅节省时间。
3.3 生成、加权、输出无偏结果
在推理阶段,流模型快速生成大量粗粒构象,然后用 PMF 计算权重并进行重加权,最后过滤掉异常样本,计算有效样本量 ESS,最终输出稳定、无偏、可用于物理分析的平衡态构象系综。
四、实验验证:精准、高效、超越传统方法
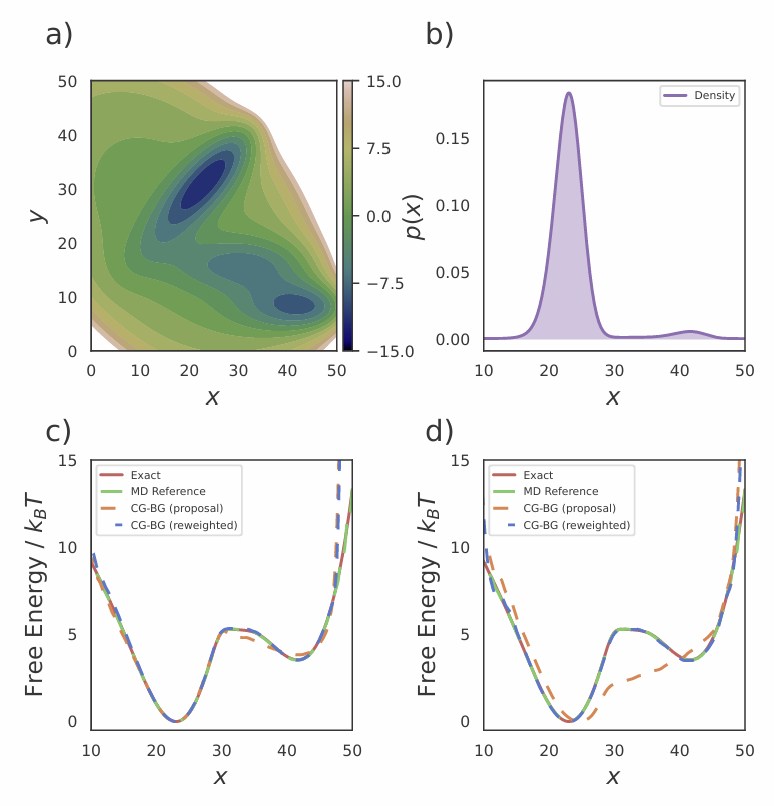
图2 Müller-Brown势能自由能曲线对比
研究团队在两类经典体系上对 CG-BG 进行了全面测试,结果极具说服力。
4.1 Müller–Brown 势能:完美恢复目标分布
在经典的二维势能面上,CG-BG 将体系投影到一维粗粒空间。实验显示,流模型本身生成的分布会偏离真实值,但经过 PMF 重加权后,几乎完美恢复理论参考分布。无论使用无偏数据还是偏置加速数据,CG-BG 都能稳定给出正确结果。
4.2 丙氨酸二肽:显式溶剂精度,粗粒度速度
在更真实的丙氨酸二肽体系中,研究团队以显式溶剂为参考基准,对比传统隐式溶剂模型。结果显示,CG-BG 预测的自由能曲线与显式溶剂几乎重合,远优于隐式溶剂。更重要的是,CG-BG 在粗粒空间中成功捕捉了溶剂介导效应,这是传统粗粒度方法几乎不可能做到的。
4.3 计算效率:速度提升超 16 倍
在速度上,CG-BG 实现了数量级的提升。粗粒度推理时间仅需 0.84 分钟 / 万样本,而全原子模型需要 13.92 分钟,速度提升超过 16 倍。训练时间也从数小时缩短到一小时以内,真正具备工业级使用价值。
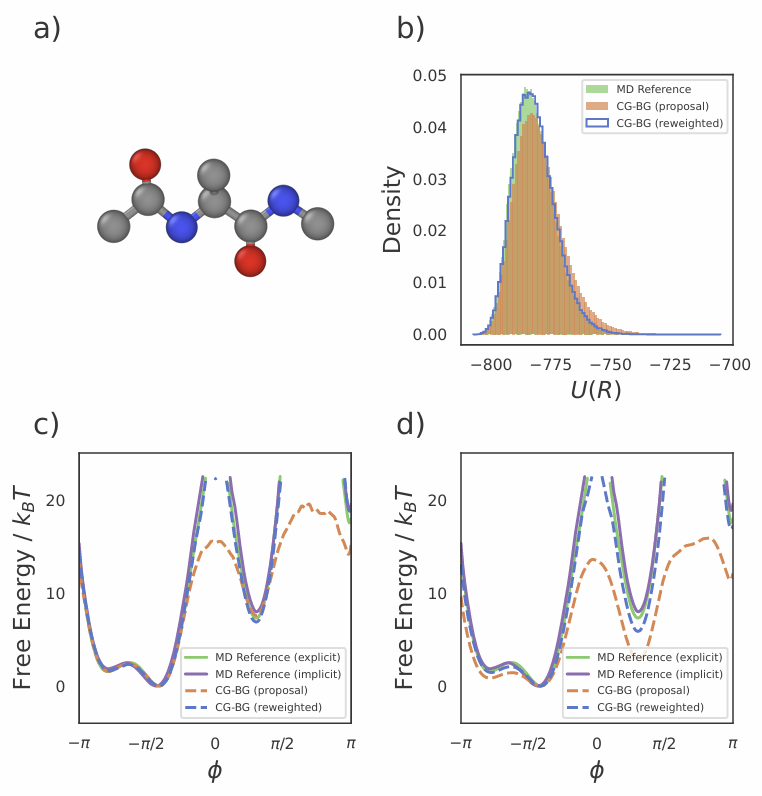
图3 丙氨酸二肽自由能
五、总结
CG-BG 的出现,标志着分子模拟进入可扩展、无偏、生成式的新时代。它解决了领域内几十年的难题:
第一,玻尔兹曼生成器无法扩展到大分子,CG-BG 用粗粒度降维解决;
第二,粗粒度模型统计有偏,CG-BG 用 PMF + 重要性采样解决;
第三,训练依赖超长平衡模拟,CG-BG 用增强采样力匹配解决。
这意味着,蛋白质构象系综、药物溶液构象、高分子聚集、膜蛋白动态等过去难以高效模拟的体系,从此可以实现快速、稳定、可信的预测。
论文链接:https://arxiv.org/abs/2602.10637
| 





















