本帖最后由 薛定谔了么 于 2025-5-28 04:10 编辑
当前模拟复杂生物系统的量子分子动力学仍受限于计算资源和方法精度,传统的DFT方法存在精度不足的问题。为此,FeNNix-Bio1的研究团队通过引入量子蒙特卡洛(QMC)和多决定因子波函数等高精度方法,构建了一个涵盖210万分子构型的大规模数据集,并成功将其用于训练FeNNix-Bio1模型,实现以接近实验的精度快速预测分子间的相互作用和反应路径。该研究不仅提高了分子模拟的精度和效率,降低了对实验的依赖,还展示了 AI 与量子化学结合的巨大潜力,为未来在药物设计、生物催化等领域的应用提供了新的工具和方法。
随着分子模拟在药物设计、生物催化和材料科学等领域的广泛应用,科学界对高精度、可扩展的模拟方法提出了更高要求。然而,传统的量子化学方法,如密度泛函理论(DFT),虽然计算效率高,但在复杂体系中的精度仍存在明显不足,对反应路径和相互作用能的精细预测的需求也难以满足。尤其在模拟生物大分子的量子分子动力学过程中,如何在保持计算可行性的同时提高模拟的准确性,一直是该领域的技术瓶颈。近年来,神经网络潜势模型的发展为原子尺度模拟提供了新的可能,但其表现高度依赖于训练数据的物理精度。因此,构建一个兼具广度与精度的大规模高质量数据集,成为推动机器学习模型走向“准实验精度”的关键步骤。
在这一背景下,Qubit Pharmaceuticals 团队联合索邦大学(Sorbonne University)推出了量子 AI 基础模型FeNNix-Bio1,学术论文《Pushing the Accuracy Limit of Foundation Neural Network Models with Quantum Monte Carlo Forces and Path Integrals》(利用量子蒙特卡罗力和路径积分突破基础神经网络模型的精度极限)展示了团队在高精度分子模拟方向的前沿探索。

几十年来,科学家们主要依赖经验力场这种基于实验数据调整的简化数学模型来预测分子的行为。尽管这类经典模型计算速度快,能够处理如蛋白质或材料等大规模体系,但它们存在显著的局限:无法准确地模拟化学反应,并且其精度高度依赖于经验参数的质量。
而如今,研究人员正逐步摆脱对传统力场的依赖。那么,这一突破是如何实现的?
答案是神经网络。它们正在被训练以“学习”量子力学规律,使其具备远超经典力场的表达能力,从而将分子模拟的精度推向新的高度。该研究的跨越式发展得益于量子化学、机器学习与高性能计算等多个领域专家的协同合作,正在重塑我们理解和建模分子世界的方式。
1. 打破传统:基础模型是一种范式转变
长期以来,研究人员在模拟分子和化学反应时,一直受限于传统的“力场”模型。这些模型依赖于固定的公式和参数,需要研究人员手动精细调整。而这种方式有一个明显的缺点:它假设原子和化学键是不会发生变化的——这显然不适合用来模拟化学反应,比如化学键断裂或新分子的形成。
但如今,借助人工智能和神经网络,研究人员找到了突破口。这些神经网络基于大量量子力学计算结果进行训练,能够跳脱传统方法的限制。因为这种新型力场是一个具备机器学习能力且能够直接理解电子的基本物理规律,所以研究团队其称为“基础模型”(Foundation Model)。
这个模型不仅能模拟简单的分子振动,还能实时追踪复杂的化学反应过程。你可以把它想象成一组“分子乐高积木”:这些积木可以根据不同的模拟需求自由组合,而不需要每次从头再来。
这种模型背后的关键优势在于其训练数据的高精度。研究团队以密度泛函理论(DFT)为基础,生成了大量分子的量子构型。虽然 DFT 相比传统力场已经更为准确,但它仍然存在一些系统性误差。于是,研究人员采用了类似“雅各布天梯”的策略,一步步向更高精度攀登。他们利用 GPU 超级计算机,选取部分分子系统,使用计算成本极高的量子蒙特卡罗(QMC)方法,甚至是多决定配置相互作用(CI)方法进行精算。这些在过去几乎只能应用于极小体系的计算,如今借助先进的超算资源,终于大规模实现——这是历史上首次在如此高的量子精度下大规模生成训练数据。
这就构成了真正意义上的“基础”模型。团队打造的第一个版本叫 FeNNix-Bio1,是专为生物分子系统设计的。它完全基于合成的量子化学数据训练而成,不依赖任何实验拟合。就像大型语言模型通过海量文本学习语言规律一样,FeNNix 则是通过海量量子计算数据,学习原子之间的相互作用规律。正因此,这种方法赋予了模型极强的通用性和稳健性。
重要的是,FeNNix-Bio1 并不局限于某一个分子或蛋白质。它是一个广泛适用的模型,未来可拓展至药物开发、催化剂设计、电池材料,甚至是核化学等多个领域。虽然目前的版本主要聚焦在生物分子上作为测试场,但名称中的 “Bio1” 已暗示了这只是一个开始。
2. 攀登阶梯:从 DFT 到 QMC
要在大规模数据集上实现量子级别的精度,并非易事。研究团队采用了多层次的计算策略,不断提升精度,这种方法也被形象地称为“攀登雅各布之梯”——这是量子化学中的经典比喻。
密度泛函理论(DFT)是目前应用最广泛的量子化学计算方法,能够覆盖大量分子结构。然而,DFT 并不是终点。为了进一步提升精度,研究团队在其中一部分代表性样本上,采用了更高级的计算方法:量子蒙特卡罗(QMC)和配置相互作用(CI)。
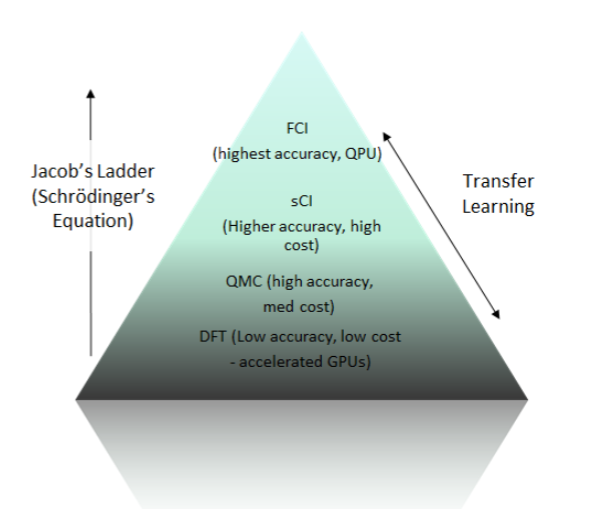
简单来说,QMC 是一种用来非常精确地求解多电子的薛定谔方程的随机方法,但是其计算量极其庞大。CI 方法则是从另一个角度出发:它考虑所有电子在所有可能状态组合中的行为,几乎能精确求解小体系的量子态。团队结合了这两种方法,用 CI 的结果引导 QMC 运算,从而获得了极高保真度的能量和力数据,作为训练基础模型的“黄金标准”。
3. 从量子精度到智能模型:让神经网络“懂”化学
在构建出高精度的量子数据集后,接下来的关键一步就是训练神经网络模型 FeNNix-Bio1。但问题来了:如何让这个模型既能继承 DFT 数据的广度,又不失 QMC 数据的精度呢?
答案是:迁移学习(transfer learning)。
这个方法既巧妙又高效。首先,研究团队用大量 DFT 数据对模型进行“基础训练”,让它掌握分子相互作用的整体趋势。然后,再用数量较少但更精确的 QMC 数据,对模型进行“精修”,让它学习 DFT 与 QMC 之间的差异,也就是所谓的“delta 校正”。
这种校正让模型在不直接看到所有 QMC 数据的情况下,依然能全面提升精度。换句话说,QMC 的“智慧”被“迁移”到了整个模型中,即使在 QMC 数据缺席的地方,也能做出接近量子级别的预测。
这也可以被比作一个学生,他先读了一本通俗易懂的教材(DFT),又听取了一位专家的精炼点评(QMC),最终他不仅知识面广,而且理解更深。研究人员在采访中也强调,这种 delta 策略是模型能够“突破 DFT 极限”而不必为每个分子配置都跑一次 QMC 的关键。最终,FeNNix-Bio1 的精度达到了远超 DFT 的水平,同时保留了速度上的优势,足以胜任日常的大规模分子模拟。
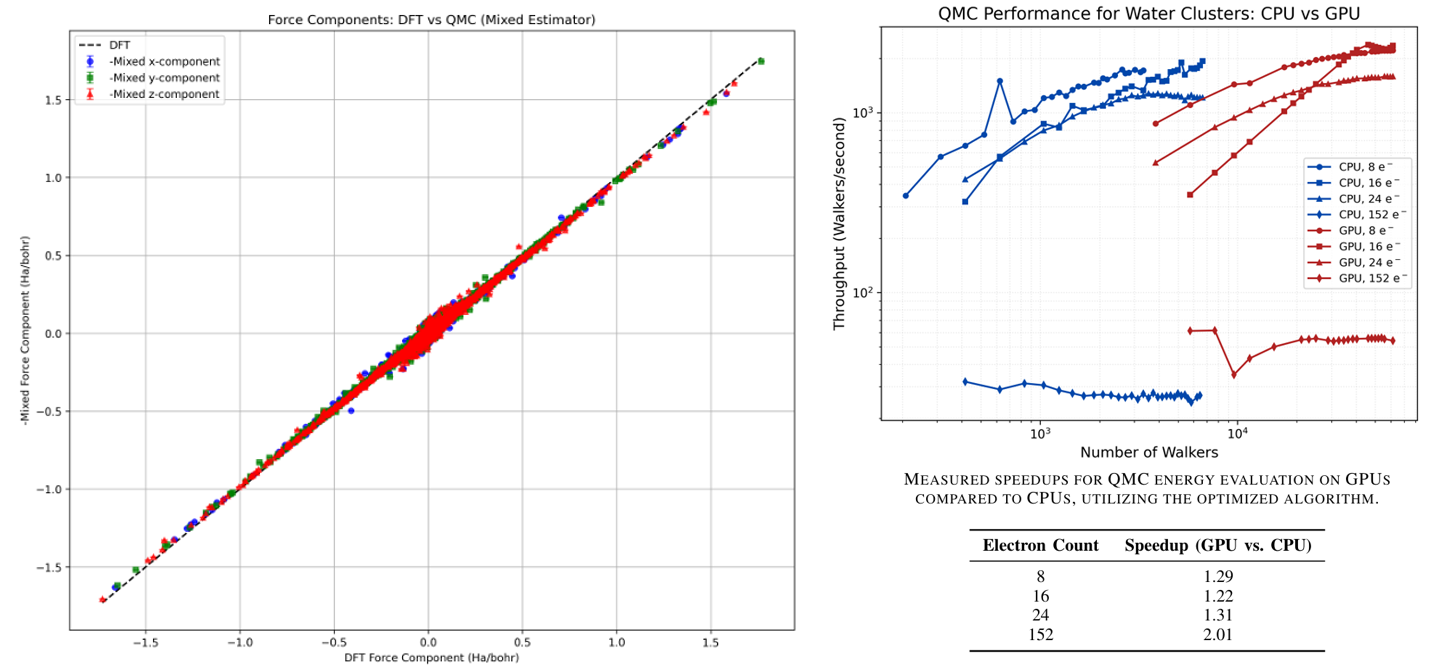
在论文中,研究团队指出,这种通过迁移学习优化 DFT 模型的方式,成功“填补了高精度量子计算与凝聚态分子动力学之间的鸿沟”。在实际应用中,FeNNix-Bio1 可以直接嵌入分子动力学模拟中,并且表现出了过去只有小体系量子模拟才能达到的真实度。
同时,作为一个“基础模型”,FeNNix-Bio1 还具备可持续升级的潜力。如果将来有了更先进的理论或与新型量子计算平台结合,团队可以直接在现有模型基础上再训练一轮,而无需像传统经验力场那样大幅修改或重写参数。这种“持续进化”的能力,正是基础模型理念最核心的价值所在。
4. 它能做的:键断裂、质子跳跃和百万原子模拟
如果这一切努力只是停留在理论层面,那也许只是学术上的“炫技”。但是,FeNNix-Bio1 已经能够运行稳定的分子动力学模拟,并且时间尺度达到数纳秒。这已经足以用来研究许多关键的化学和生物过程,比如蛋白质折叠。而它更令人兴奋的突破是它可以模拟化学键的断裂和形成,这是传统力场完全做不到的,因为它们的公式结构不允许原子“重新组合”。
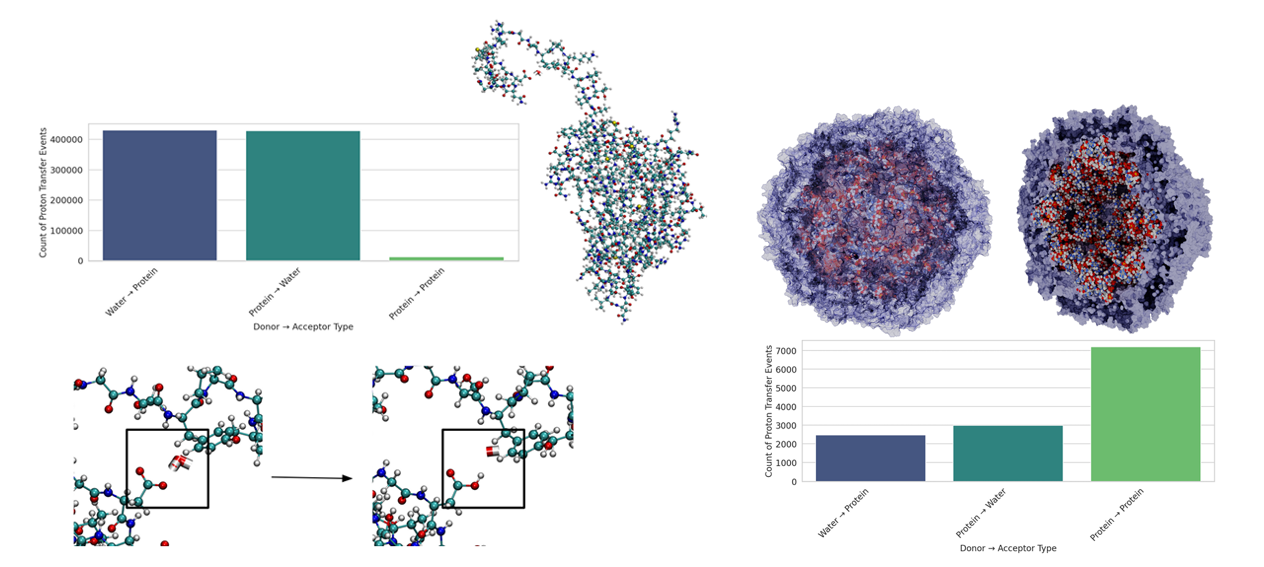
FeNNix-Bio1 做到了这一步。它基于物理法则,可以自然地模拟化学键的断裂与生成。研究团队特别演示了质子转移反应的模拟,也就是一个质子在不同原子之间“跳跃”。这种反应在酶的活性、药物的作用机制,甚至辐射对 DNA 的破坏中都非常关键。而现在,这些过程可以被大规模、可信度高地模拟出来。
此外,团队进行了一个几年前几乎无法想象的模拟——他们选取了完整包水的烟草花叶病毒(约 100 万个原子),在追踪质子的量子隧穿效应的同时,还在模拟中重现了复杂的化学相互作用,例如质子化状态的变化和化学键的调整。该环境涵盖了真实病毒本体、其 RNA 遗传物质、水分子以及离子,构成了一个真正复杂的生物系统。
5. 迎接模拟新时代:从经验力场迈向科学智能
总的来说,该研究不只是一个小的改进,它彻底打破了“精度与规模不可兼得”的旧范式,展现出只要在前期投入算力,用最好的量子数据训练出一个基础模型,就能收获一个可以处理极其复杂分子现象的全能工具。
这标志着一种潜在的新模式正在到来:在计算机上进行的“in silico”实验,未来可能与传统的“in vitro”(体外实验)并驾齐驱,甚至在某些场景中取而代之。研究人员能更有信心地用模拟结果指导假设验证、设计实验、加快研发。
6. 未来展望:基础模型与光量子计算机的协同进化
FeNNix-Bio1 展示了人工智能与量子化学结合所能达到的全新高度。它通过深度神经网络学习大量量子力学数据,成功突破了传统分子模拟在精度与规模之间的权衡。而展望未来,这一类“基础模型”与光量子计算机的结合,将进一步释放其潜力。
光量子计算机擅长处理高维、并行的量子态演化问题,尤其在模拟电子结构、核量子效应、强关联体系等领域具有独特优势。未来,我们可以利用光量子计算平台生成更高精度、更复杂体系的参考数据,作为训练或微调基础模型的关键资源;也可以借助其原生的量子动力学模拟能力,辅助模型准确处理路径积分、质子跳跃等现象。
从长远看,这种协同将催生一种闭环式的“量子增强模拟”架构:神经网络模型提供初步预测与结构筛选,光量子计算进行关键态点的精算反馈,持续优化模型性能与适应性。这不仅能够推动分子模拟走向更高的精度、更大的系统和更复杂的反应,也将成为跨学科科学协作的新典范。
在模拟生命分子机器、设计新能源材料、探索极端化学空间的征途中,AI 与光量子计算机的融合,或许就是我们打开下一个科学时代之门的钥匙。
本文改编转载自Qubit Pharmaceuticals 官方博客发布的文章。
原论文链接:https://arxiv.org/abs/2504.07948 | 





















