本帖最后由 graphite 于 2025-6-16 17:23 编辑
人工智能(AI)正在彻底改变科学家预测蛋白质相互作用的方式。以前的方法又慢又不准,而AI能直接从蛋白质序列一口气预测整个复合体的结构,精度大幅提升。不过,AI目前还有几大挑战:难以预测结合时蛋白质的结构变化、依赖可能不足的进化信息、难以处理没有固定形状的蛋白质区域,以及预测大型复合物很吃力。未来需要结合更创新的混合方法、更深度的数据整合以及更强大的下一代工具,才能克服这些难题,更好地助力新药研发和理解生命活动。
2025年6月,来自华中科技大学和华沙大学生物化学研究中心的研究人员在Drug Discovery Today上发表文章Computational methods for modeling protein – protein interactions in the AI era: Current status and future directions,主要探讨了在人工智能时代下,计算方法在蛋白质-蛋白质相互作用(PPI)建模中的现状与未来发展方向。

长久以来,蛋白质-蛋白质复合物结构的计算预测主要依赖蛋白质-蛋白质对接技术。这种方法通常分为两大类:模板依赖型对接和模板无关型对接。前者在存在高度同源复合物模板(如来自蛋白质数据库PDB)时效果显著(例如PRISM,HMI-PRED 等方法);后者则在缺乏模板时,通过探索巨大的构象空间来寻找可能的结合模式(例如HDOCK, ZDOCK, ClusPro, GRAMM, PatchDock, LzerD, HADDOCK, RosettaDock, SwarmDock, LightDock等)。然而,传统对接方法,尤其是模板无关型对接,长期受限于采样效率和评分函数的准确性,特别是在处理蛋白质柔性时面临巨大挑战。
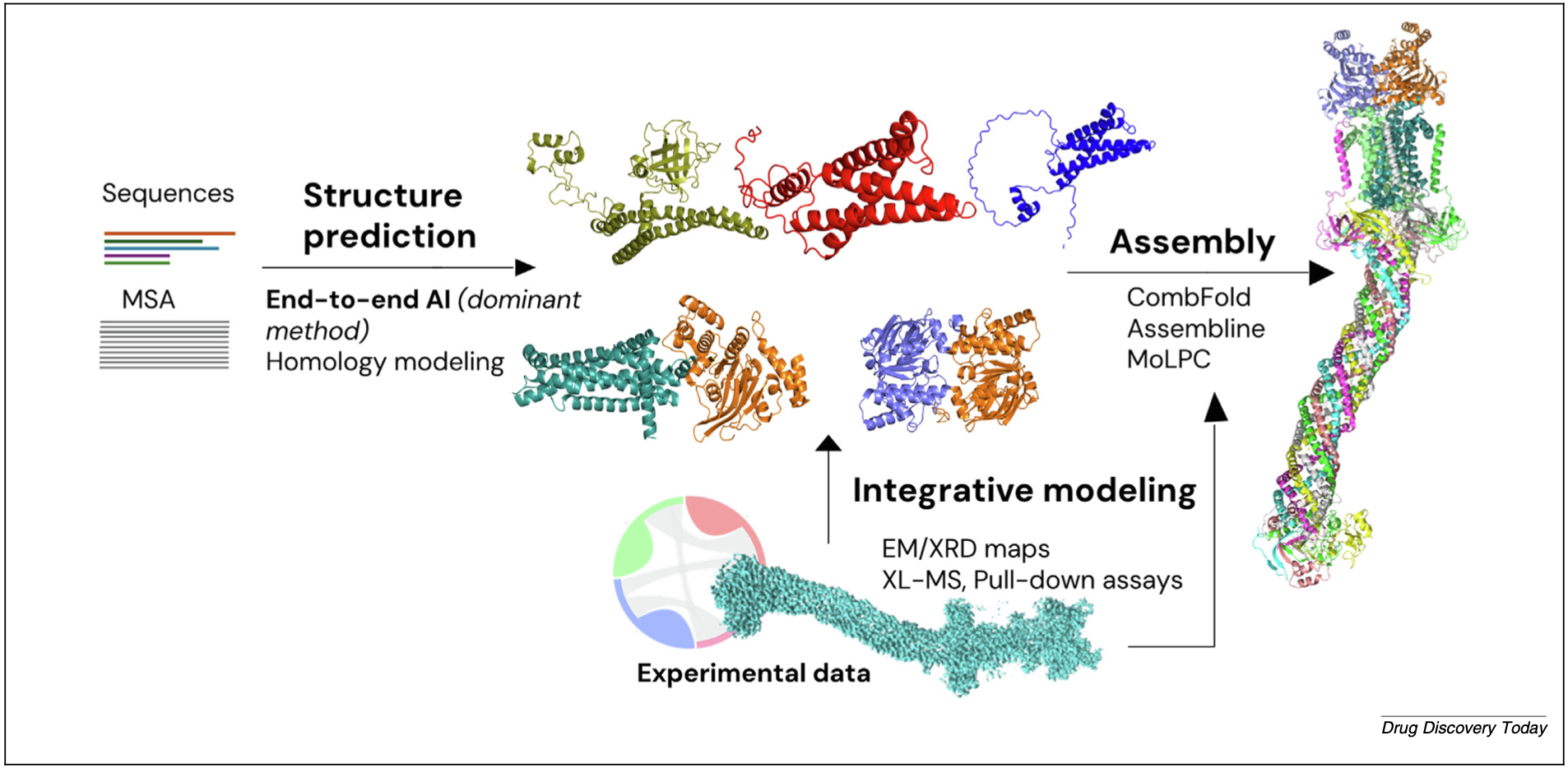
图1 人工智能时代蛋白质-蛋白质复合物建模的通用流程
近年来,以AlphaFold(AF)及其衍生技术(特别是AlphaFold-Multimer(AF-Multimer)和最近的AlphaFold 3(AF3))为代表的端到端深度学习框架彻底改变了这一领域。这些模型能够直接从蛋白质序列(及多序列比对MSA数据)出发,同时预测整个复合物的三维结构,绕过了传统对接中分离的采样和评分步骤,实现了前所未有的预测精度。它们强大的核心在于利用大规模数据集和神经网络直接推断残基-残基接触(Residue-Residue Contacts)和整体结构构型。尽管对于大型组装体,目前实践中常采用“先预测单体亚基结构,再计算组装成全复合物”的两步流程(如使用CombFold,Assembline,MoLPC等工具),并可通过整合冷冻电镜(Cryo-EM)、X射线衍射(XRD)、交联质谱(XL-MS)等实验数据进行优化(如图1所示),但AI驱动的端到端预测已成为当前PPI结构预测的主导范式。
尽管端到端AI模型展现出强大的性能,传统对接方法并未完全退出舞台。它们在特定场景下(如当存在可靠模板或需要快速筛选时)依然具有价值。更重要的是,AI技术本身也在赋能传统对接方法。
在采样方面,除了传统基于几何互补性、局部形状匹配或启发式搜索的算法,新兴的深度学习对接方法(如EQUIDOCK,DIFFDOCK-PP,DockGPT,GeoDock)利用神经网络学习结合模式,实现了更快的推理速度,尽管目前其绝对精度通常低于端到端方法。
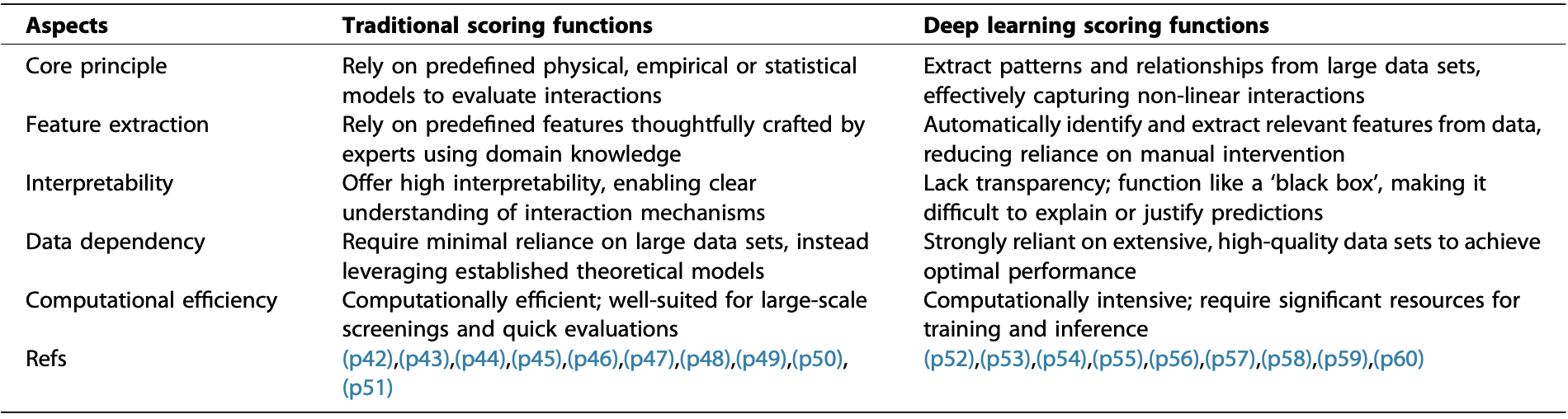
在评分环节,AI的贡献尤为显著。传统的评分函数依赖于预定义的物理模型、经验模型或统计模型(如HawkRank,SDOCK,pyDock,ITScore-PP,ZRANK,FireDock,SIPPER,DECK等),它们计算效率高、可解释性强。而深度学习评分函数(如TRScore,DeepRank,DeepLoc,GNN-DOVE,DeepRank-GNN, DeepRank-GNN-esmBA,G-RANK,PInet等)则能够从海量数据中自动提取特征,捕捉复杂的非线性相互作用模式,显著提升了区分近天然构象的能力。当然,深度学习评分函数依赖高质量大数据且计算成本较高、可解释性相对较弱。综述指出,结合传统和深度学习评分函数的优势,可能是未来开发更鲁棒、更准确对接工具的方向(如表1所示)。
表1 传统评分函数与深度学习评分函数的对比
鉴于AlphaFold2(AF2)在单体蛋白质结构预测上的革命性成功,研究者们积极探索将其应用于复合物预测。早期策略包括将不同蛋白链的氨基酸序列用多甘氨酸连接子拼接成一条“伪序列”输入AF2。随后,专门为复合物预测设计的AF-Multimer及其后续改进版本,通过显式建模链间相互作用,显著提升了异源多聚体预测的准确性。最新的AF3更进一步,不仅预测蛋白质-蛋白质复合物,还能预测涉及蛋白质与核酸、小分子配体等更广泛生物分子相互作用的复合物结构,这对于理解许多关键生物过程和药物设计至关重要。
然而,AlphaFold家族方法也存在其局限性。它们高度依赖共进化信号(主要通过MSA获取)。对于进化信号弱(如弱相互作用、瞬时相互作用)或缺乏足够同源序列的蛋白质对,其预测性能会显著下降。此外,AF3虽然通过公共服务器提升了可及性,但其巨大的计算资源需求仍限制大规模或本地部署,简化版如ColabFold虽降低了门槛,但存在计算限制等实际问题。
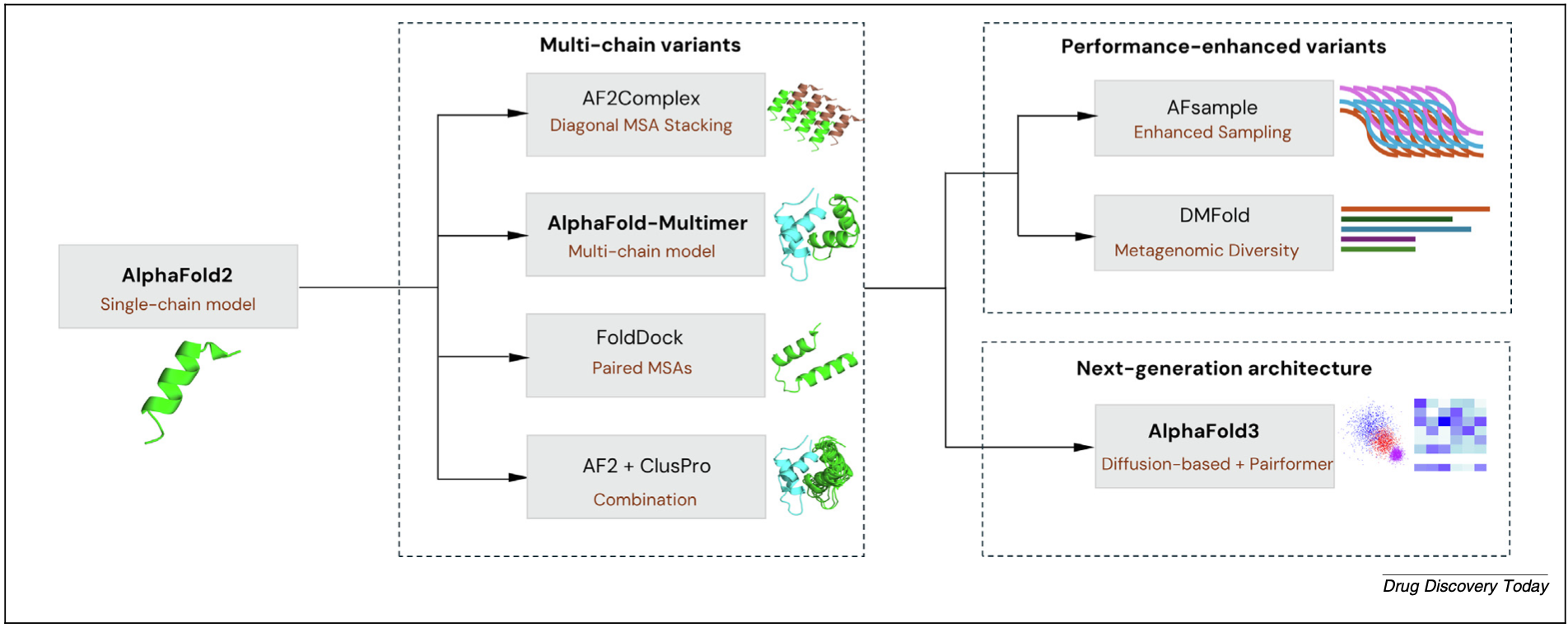
图2 基于AlphaFold的蛋白质复合物结构预测方法的演进
尽管AI带来了巨大进步,PPI结构预测仍面临几个关键的科学挑战:
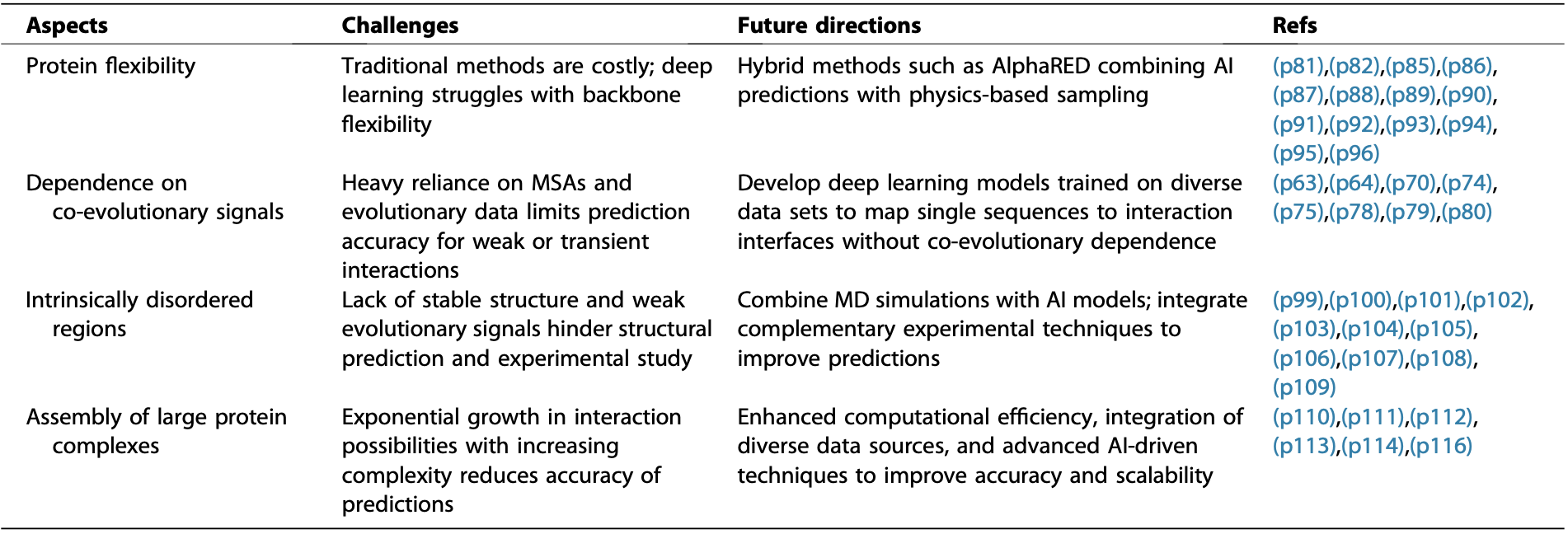
蛋白质柔性:蛋白质在结合过程中常发生构象变化(诱导契合,Induced Fit)。传统方法模拟这种柔性(如分子动力学MD模拟、旋转异构体库、弹性网络模型ENMs、粗粒化模型)计算成本高昂且精度有限。AF-Multimer等AI方法在预测结构良好的复合物上表现出色,但对涉及显著构象变化的体系(如抗体-抗原)仍力有不逮。新兴的混合方法(如AlphaRED)结合AI预测与基于物理的采样,以及利用AI生成分子轨迹或结合流模型将静态预测转化为动态系综,展现出解决这一难题的潜力。
共进化信号依赖:如前所述,当前最先进的端到端模型严重依赖MSA中的共进化信息来推断相互作用界面。开发能够仅从单序列出发预测相互作用界面的深度学习模型,是克服这一局限、拓展预测范围的关键方向。
内在无序区域(IDRs)的建模:许多蛋白质包含缺乏稳定三维结构的无序区域(IDRs),它们在信号传导等过程中通过动态相互作用发挥关键功能。然而,IDRs缺乏稳定的结构特征和强的进化信号,使得其结构预测和相互作用研究极其困难。解决之道在于整合分子动力学(MD)模拟与AI模型。例如,利用大规模MD数据训练AI模型(如ALBATROSS),或利用AI预测(如AF2)为无序蛋白的MD模拟提供距离约束(如AlphaFold-Meta推断方法),形成迭代优化的闭环。互补性实验技术(如NMR)的整合也至关重要。
大型蛋白质复合物的组装:随着复合物规模增大,亚基间可能的相互作用方式呈指数级增长,导致预测精度下降和计算复杂度激增。提高计算效率、整合多种数据源(Cryo-EM,XL-MS,Pull-down assays)以及开发更先进的AI驱动组装算法(如CombFold,Assembline,AlphaPulldown)是提升大型复合物建模准确性和可扩展性的关键。
表2 蛋白质-蛋白质相互作用建模的挑战与未来方向
人工智能,特别是深度学习,已经并将持续重塑PPI计算建模的格局。以AlphaFold及其衍生技术为代表的端到端框架,在预测精度和效率上实现了质的飞跃,极大地推动了我们对生物分子相互作用机制的理解。然而,蛋白质柔性、共进化信号依赖、无序区相互作用以及大型复合物组装等挑战依然存在,呼唤着更创新的混合方法、更深度的数据整合以及更强大的下一代AI工具。
这些计算方法的进步正在产生切实的影响:从de novo设计高亲和力的迷你蛋白结合剂,到开发免疫治疗新策略(如抗体设计),再到自动化设计功能蛋白结合剂平台(如BindCraft)的出现,都彰显了计算模型在生物技术和生物医学研究中日益增长的核心作用。我们有理由相信,随着计算生物学、人工智能和实验技术的深度融合与持续创新,蛋白质相互作用的结构预测与功能解析将迎来更加辉煌的未来,为生命科学研究和精准药物设计开辟更广阔的道路。
展望未来,克服现有挑战需要多方面的协同努力:
发展混合建模策略:将深度学习预测与基于物理的模拟(如MD)和动态采样方法相结合,以更有效地捕捉蛋白质的构象动态和复杂的相互作用能量景观。
深度整合多源实验数据:Cryo-EM、XL-MS、Pull-down等实验数据对于验证、优化和约束计算模型不可或缺,是实现高精度预测的基石。方法如AF3整合交联数据(Crosslinks)即是有益的尝试。
构建可扩展、泛化性强的AI框架:探索利用基础模型(Foundation Models)、多任务学习(Multi-task Learning)等策略,开发计算效率更高、适用范围更广、鲁棒性更强的预测工具。
平衡精度、效率与可及性:在追求更高预测精度的同时,必须考虑计算成本和工具的易用性,确保先进方法能够惠及更广大的生物医学研究群体。
| 





















