本帖最后由 薛定谔了么 于 2025-10-17 21:40 编辑
随着计算能力的显著提升以及分子动力学模拟软件的不断进步,如何高效地存储和共享庞大的生物分子模拟数据集已成为一项关键挑战。对此,塔塔基础科学研究所以及波鸿鲁尔大学研究人员于2025年8月18日在《Journal of Chemical Information and Modeling》上发表文章,题为“Employing Artificial Neural Networks for Optimal Storage and Facile Sharing of Molecular Dynamics Simulation Trajectories”。

作者开发出了一种能够将MD轨迹压缩至极小潜在空间的方法。与广泛使用的MD程序包GROMACS中的高压缩轨迹格式xtc相比,该方法可节省高达98%的磁盘空间,从而极大地促进了模拟轨迹的存储与共享。该方法为科研界高效存储和共享大规模生物分子模拟数据提供了可能,从而有望促进协同研究。完整的工作流程称为compresstraj。
代码仓库:https://github.com/SerpentByte/compresstraj
背景
分子动力学(MD)模拟在探索和理解复杂生物分子体系中的应用日益广泛。MD轨迹,尤其是针对大型模拟体系(如生物大分子)时,往往会产生从数百GB到数十TB的庞大数据量。这类数据的存储与共享自然面临重大挑战。由于参与MD模拟的研究人员在研究目标、视角和对体系的理解方面各不相同,模拟轨迹中蕴含的大量潜在信息往往未被充分利用。若能有效地分发这些数据集,将有助于多角度的深入探索,从而提高知识挖掘的效率。生物分子模拟领域已从诸如PDB和SwissProt等成熟数据库中获益良多。然而,目前仍存在显著的空白,即缺乏一个用于统一存储与共享模拟轨迹的数据库。
在本研究中,作者采用自编码器(AE)对分子动力学轨迹的每一帧进行编码,将其映射至紧凑的潜在空间表示,并视其为压缩后的形式。自编码器在生物分子模拟中的应用主要集中于将高维数据(如距离矩阵)投影到低维潜在空间。然而,直接以坐标作为输入的做法较为罕见,这是因为坐标缺乏三维特殊欧几里得群的不变性,即使对整个体系进行旋转或平移,体系的本质特征不变,但坐标值却会不同。作者直接使用原子坐标来训练自编码器,并将其潜在空间作为压缩轨迹。结果表明,潜在空间能够高度压缩MD轨迹,并在解压后准确地重建粒子坐标。这使得生成的高压缩MD轨迹不仅易于存储与共享,而且在需要时可快速解压。重要的是,机器学习重建的轨迹在关键结构特征方面与原始轨迹高度一致。
方法
本研究采用的生物分子体系如图1和表1所示,这些体系在尺寸与柔性方面覆盖了广泛的范围,从柔性的短肽(如chignolin),到折叠的迷你蛋白/球状蛋白(如trpcage、HEWL),再到天然无序蛋白(Aβ40、ACTR)、脂质膜体系(DOPC双分子层)、蛋白-配体相互作用体系(T4L L99+苯、Cyt-P450+莰酮)、含有修饰氨基酸的大型蛋白复合物(SARS-CoV-2刺突蛋白)、膜蛋白体系(CymA-PHPC膜)以及溶剂体系(TIP3P水)。这些体系的选择基于其具备的长时间尺度模拟轨迹,数据部分由D. E. Shaw Research慷慨提供,部分来自公开数据库,另有部分由该研究组自行生成。
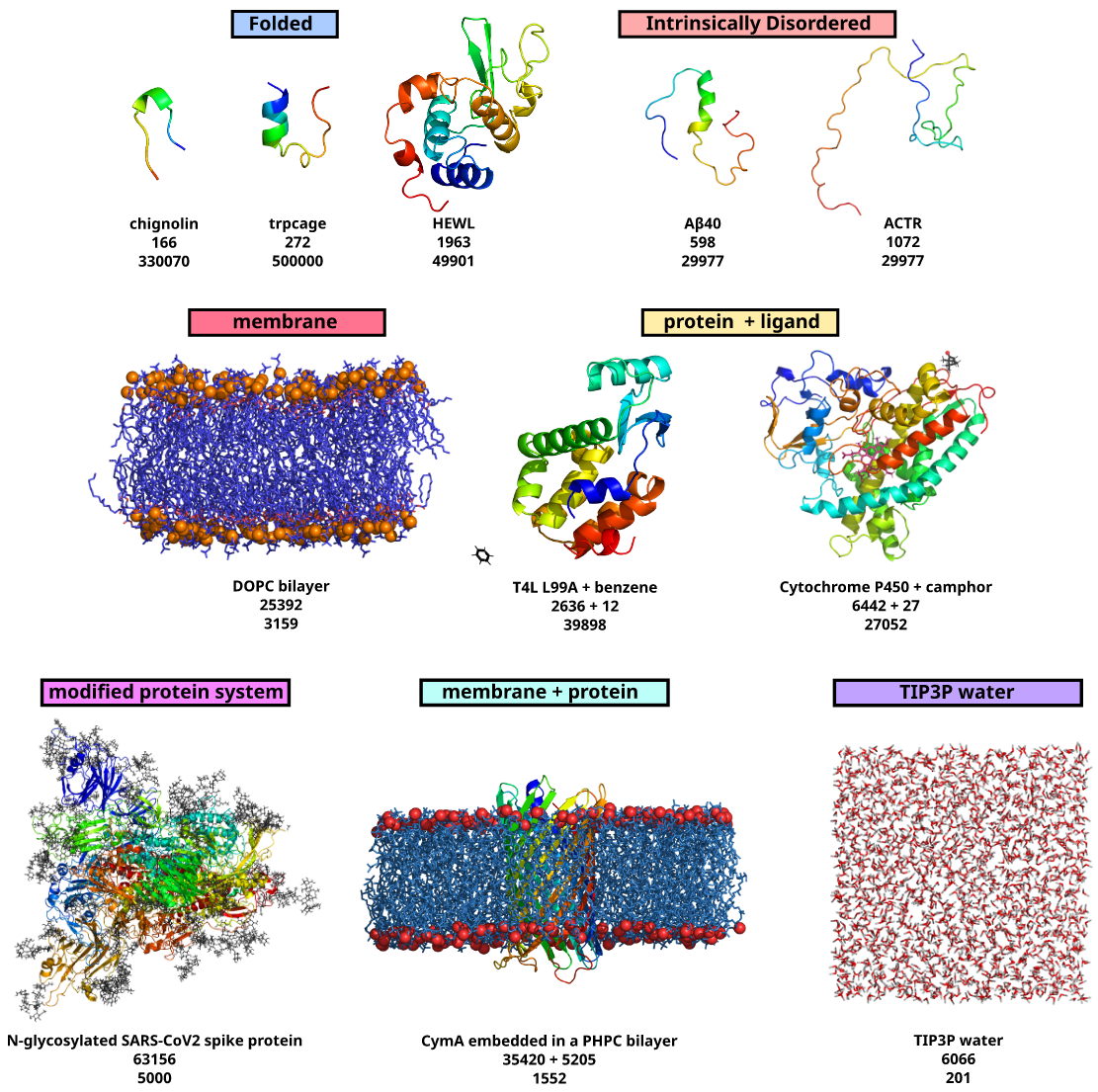
图1 研究所使用体系的代表性构象,中间数字为原子数,下方数字为各轨迹的帧数
对于轨迹的处理,该流程的第一步是从轨迹中选取一个帧作为参考构象。具体选择哪一帧是任意的,对最终结果无影响。轨迹首先与参考帧进行对齐处理,随后,对于对齐后的每一帧,将体系的质心平移至坐标原点。这些变换使所得坐标具备伪SE(3)不变性,从而可作为AE的输入。对于含有N个粒子的体系,其轨迹包含3N个笛卡尔坐标分量。在训练AE前,使用min-max缩放器对各维度坐标进行缩放,将最小值设为0、最大值设为1。每一帧的坐标矩阵维度为(N, 3),在展平后变为一个长度为3N的向量。所有帧的数据随后汇总,得到一个维度为(f, 3N)的数据集,其中f为轨迹帧数。最终,缩放后的轨迹作为AE的输入进行训练。
表1 研究中使用的不同模拟体系与MD轨迹的详细信息
本研究中使用的自编码器架构在所有体系中保持一致(图2)。采用了全连接型自编码器,即网络仅由全连接的前馈层组成。除输出层外,所有层均采用ELU激活函数,输出层则使用sigmoid激活函数。模型的损失函数为RMSE,优化器使用AdamW,每个模型至少训练2000个epoch。
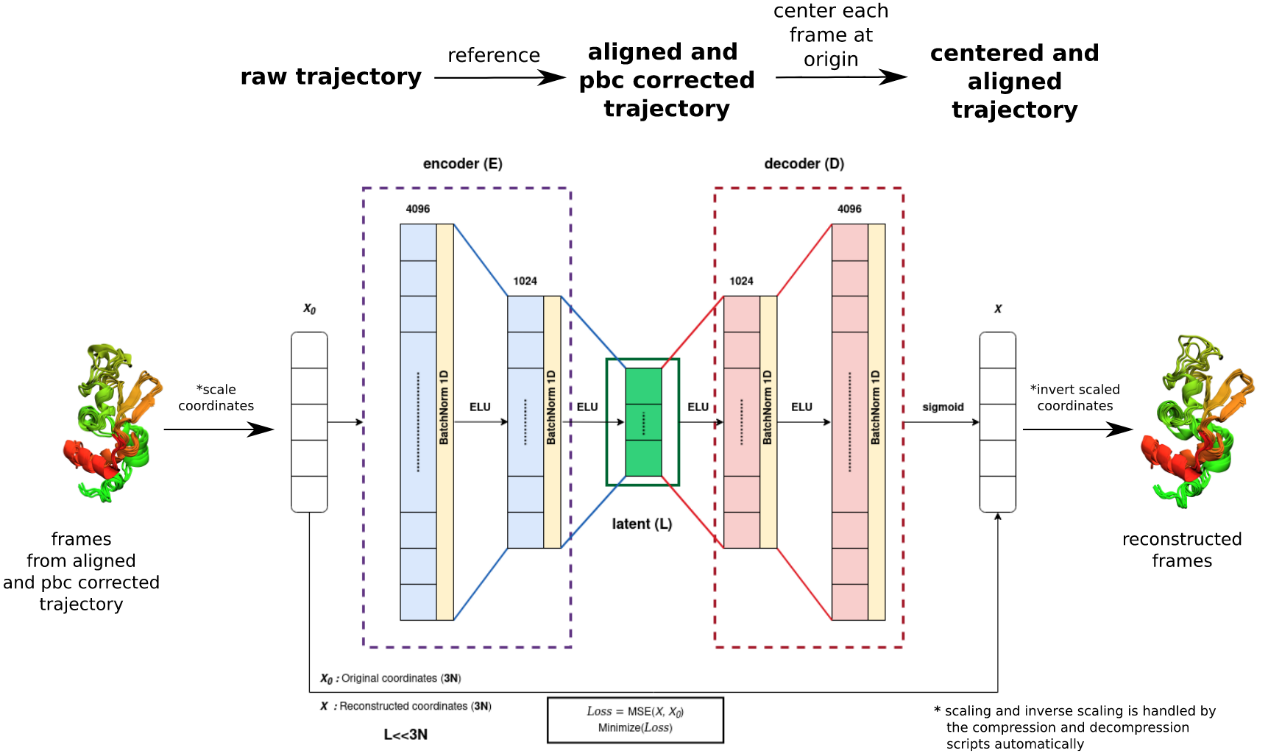
图2 研究中使用的自编码器架构示意图
为便于轨迹的预处理与自编码器的训练,开发了一个名为compresstraj的Python库,该仓库提供了可直接使用的Python脚本,一个用于压缩、一个用于解压缩,另一个用于重组分片化的体系。模型训练完成后,通过自编码器的编码器部分将坐标投影至潜在空间,即可获得压缩后的轨迹。在解压过程中,使用自编码器的解码器部分将潜在表示解码为缩放后的坐标。由于算法与输入体系的完整性无关,因此任何体系都可以拆分为多个片段,每个片段作为独立单元进行压缩与解压。各片段独立压缩、解压后,可使用脚本重新组合,恢复完整轨迹。表2、3给出了计算资源使用情况。
表2 不同体系规模下训练所需GPU显存使用情况
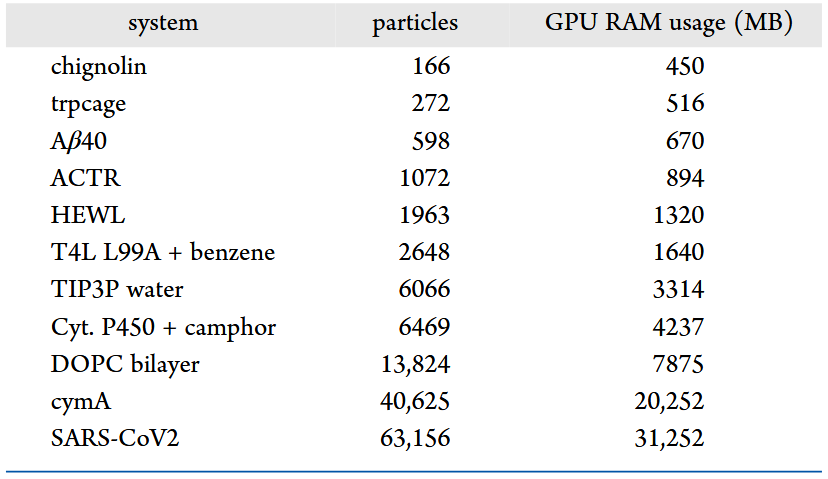
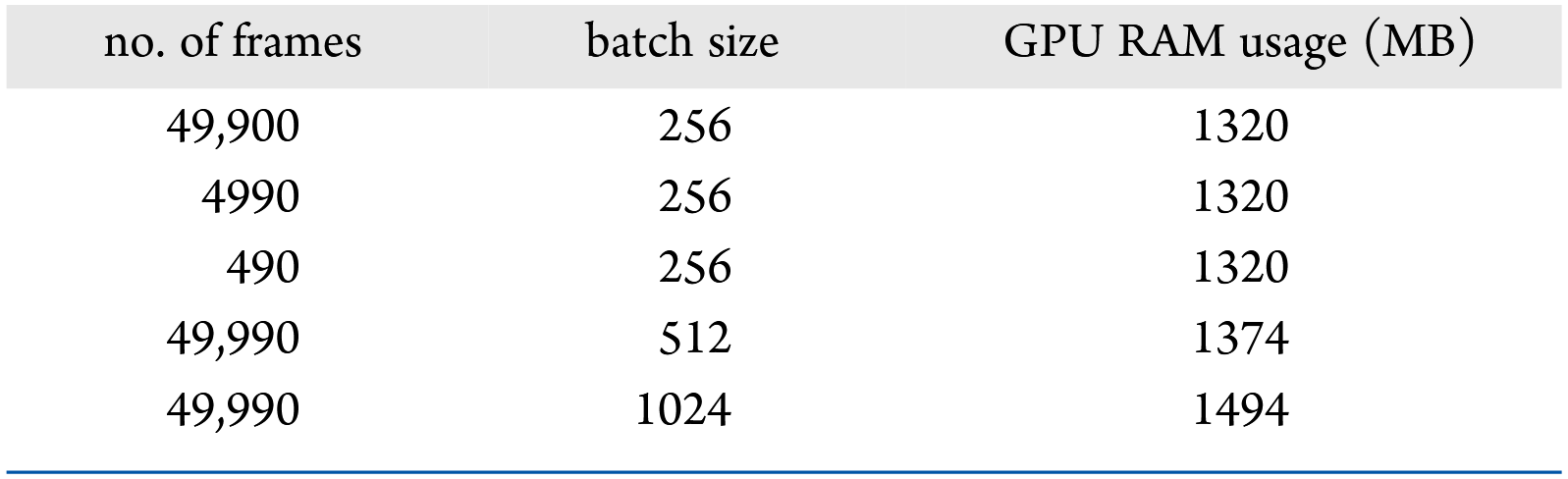
表3 在不同轨迹长度与批量大小下HEWL体系的GPU显存使用情况
结果
压缩HEWL轨迹及其重建
首先在卵清溶菌酶(HEWL)(PDB ID:1AKI)体系上开展了基于自编码器流程的验证研究。在包含49901帧的轨迹上训练了自编码器,该轨迹记录了包括氢原子在内的所有HEWL原子的三维笛卡尔坐标。如图3a所示,训练损失在初期迅速下降,随后趋于平稳。原始构象与重建构象间的RMSD分布非常集中(图3b),这表明从压缩轨迹中重建的原子坐标精度极高。比较了原始轨迹(图3c)与重建轨迹(图3d)所得的Ramachandran图。正如较小RMSD值所预示的那样,分析结果表明轨迹从潜在空间重建后其二级结构保持完好。
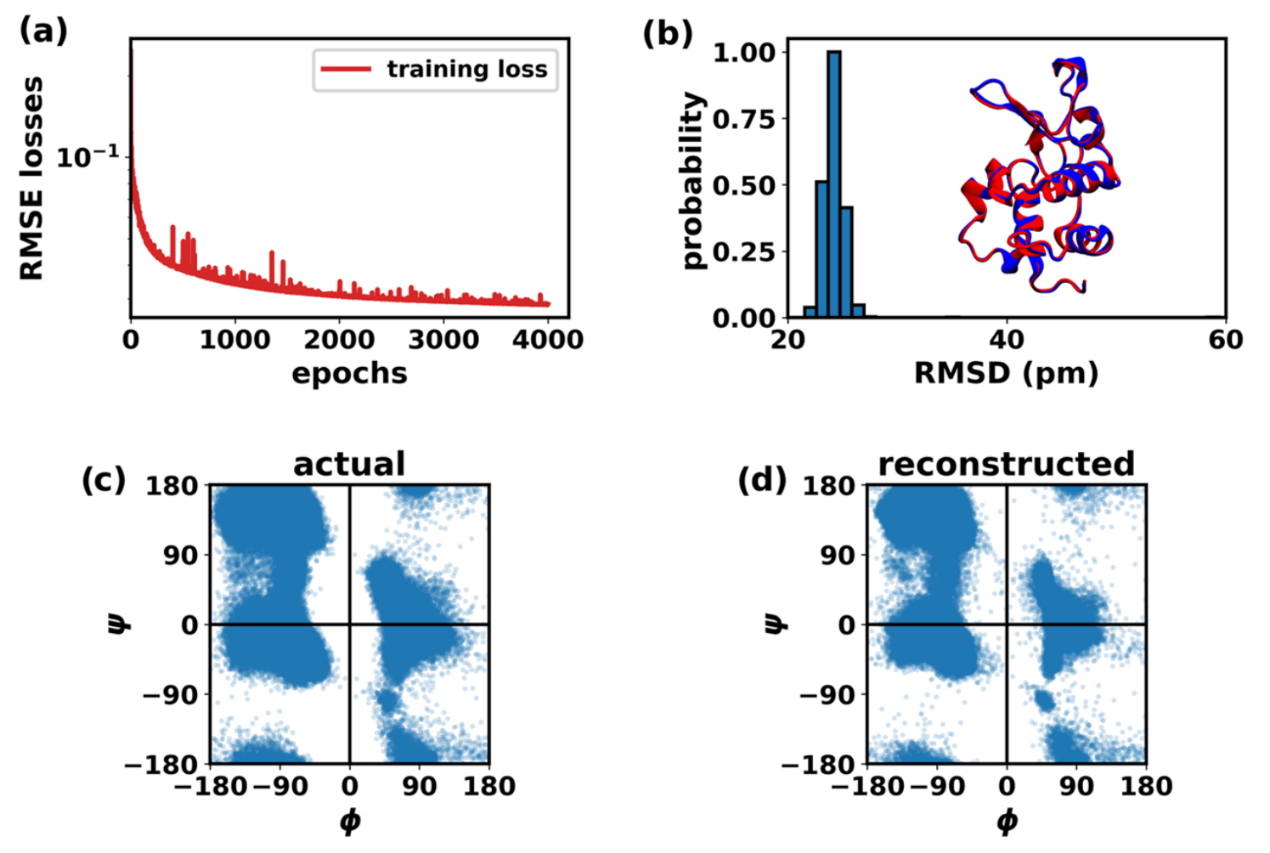
图3 HEWL体系在压缩参数c=20下的压缩与重建结果
在多种蛋白质体系上评估
图4a、4b展示了训练与验证损失结果,表明模型能够基于已对齐、居中且缩放的坐标高效训练。结果显示,折叠蛋白质体系的重建精度略高于固有无序蛋白(IDPs)。这种差异可能源于IDPs体系中更显著的构象波动,它们会影响轨迹相对于参考构象的对齐,从而削弱其伪SE(3)不变性。评估二级结构的重建效果,计算了原始与重建残基—残基间Cα接触图的MAE(图4d)。如前述的亚埃级重建精度所预示,MAE值极低,说明在解压后主链骨架结构能够被准确恢复。
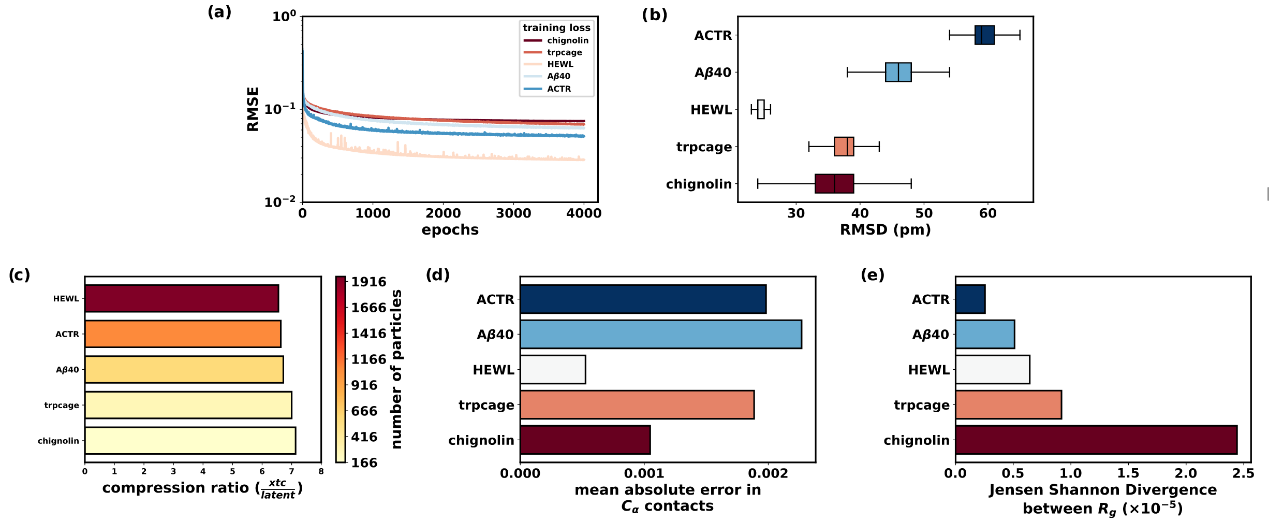
图4 折叠蛋白与天然无序蛋白的压缩结果
扩展至膜体系
将该方法扩展至由多种脂质分子组成的膜体系。图5a显示了解压后帧(红色)与原始帧(蓝色)的叠合结果。在整个轨迹中,帧间RMSD范围为41-88pm,表明系统被高精度地重建。为进一步验证重建膜体系的可靠性,分析了原始与重建轨迹的SCC键序参数(图5b)。结果表明,重建轨迹中的有序度参数与原始数据高度一致。综上,该算法能够有效压缩磷脂双分子层的分子动力学轨迹,并在解压后准确重建原始体系结构与性质。
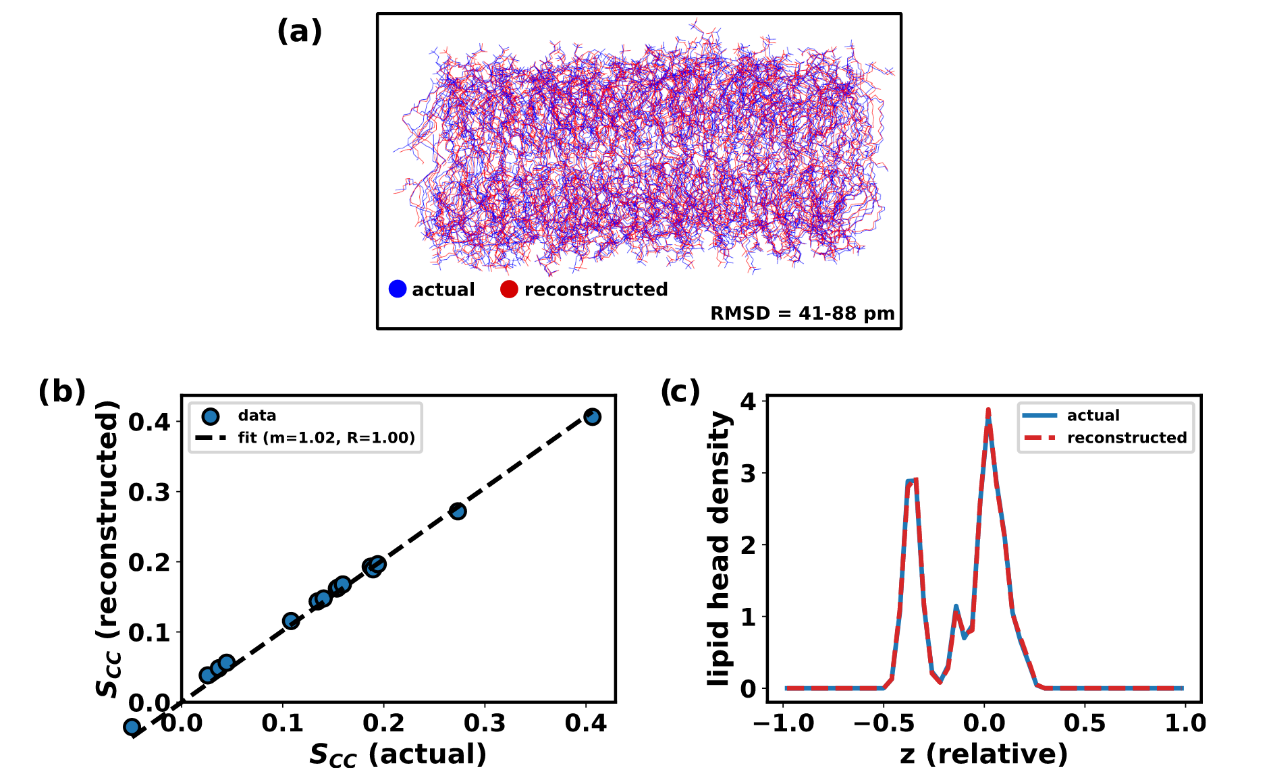
图5 DOPC双分子层的压缩与解压结果
多组分复合体系的测试
许多实际研究涉及更复杂的体系,例如蛋白-配体复合物、蛋白-蛋白复合物以及膜结合蛋白。进一步展示AE算法在这类多组分复杂体系中同样可以被可靠地应用,并能准确重建与功能相关的关键性质。
对于蛋白-配体体系,选择了两个典型体系作为测试案例,T4L L99A突变体与苯结合体系以及细胞色素P450与莰酮结合体系。按照前述流程对两条轨迹进行了压缩与重建。训练损失与帧间RMSD的结果均表明重建精度极高(图6a,b)。当蛋白部分采用c=20,配体部分采用L=4时,两体系均实现了约85%的压缩率(图6c)。残基间Cα接触图的MAE(图6d)进一步验证了结构的准确重建。
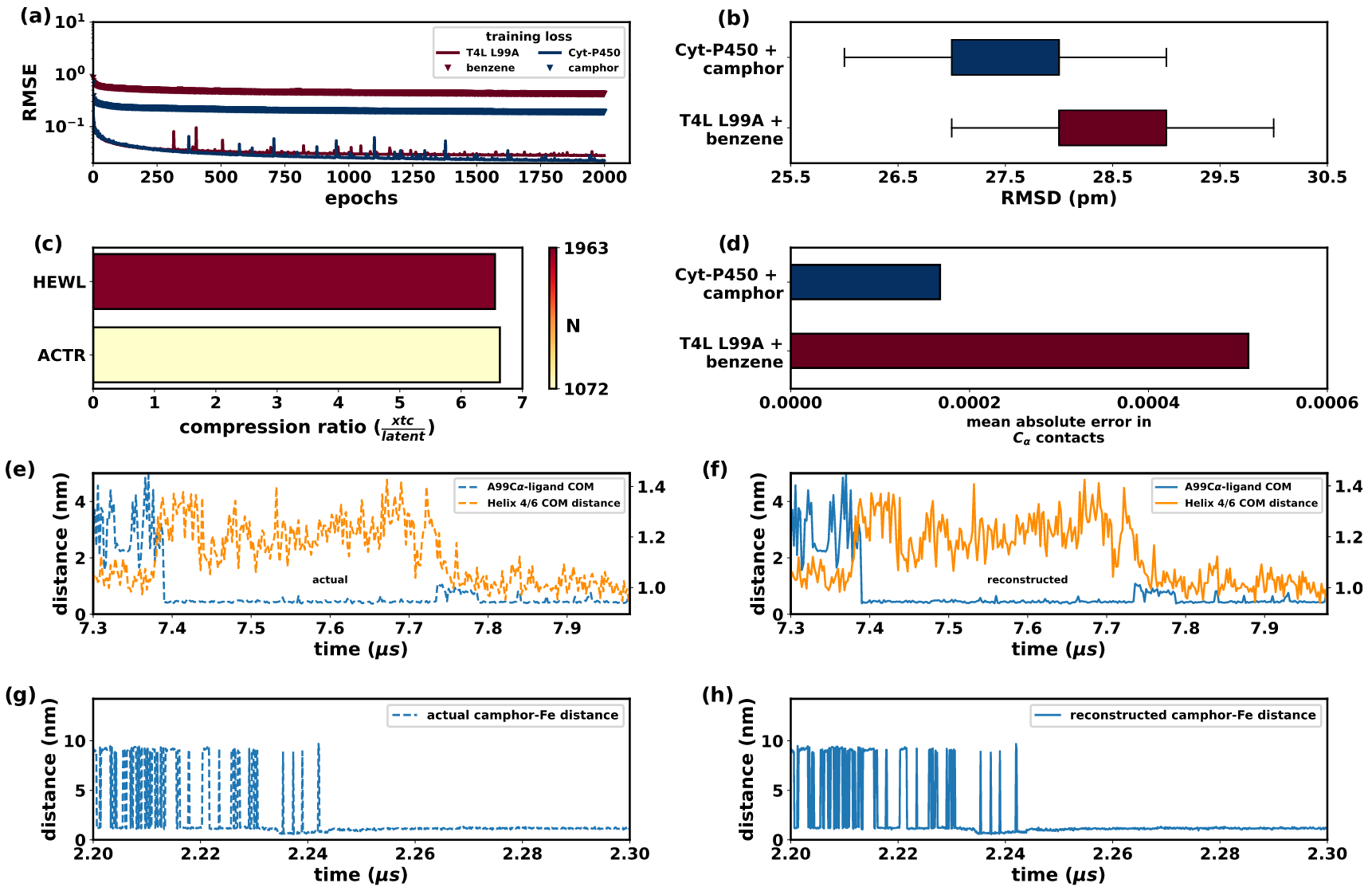
图6 蛋白-配体体系的压缩结果
进一步验证该方法在大型糖基化复合体系中的适用性,选取了SARS-CoV-2刺突糖蛋白的头部区域作为测试对象。图7a展示了解压后帧(红色)与原始帧(蓝色)的叠合;图7b为糖链区域的局部放大图。这些结果表明,该算法在大型复杂体系(如糖基化蛋白-蛋白复合物)中同样能够实现高压缩率与高精度的重建。
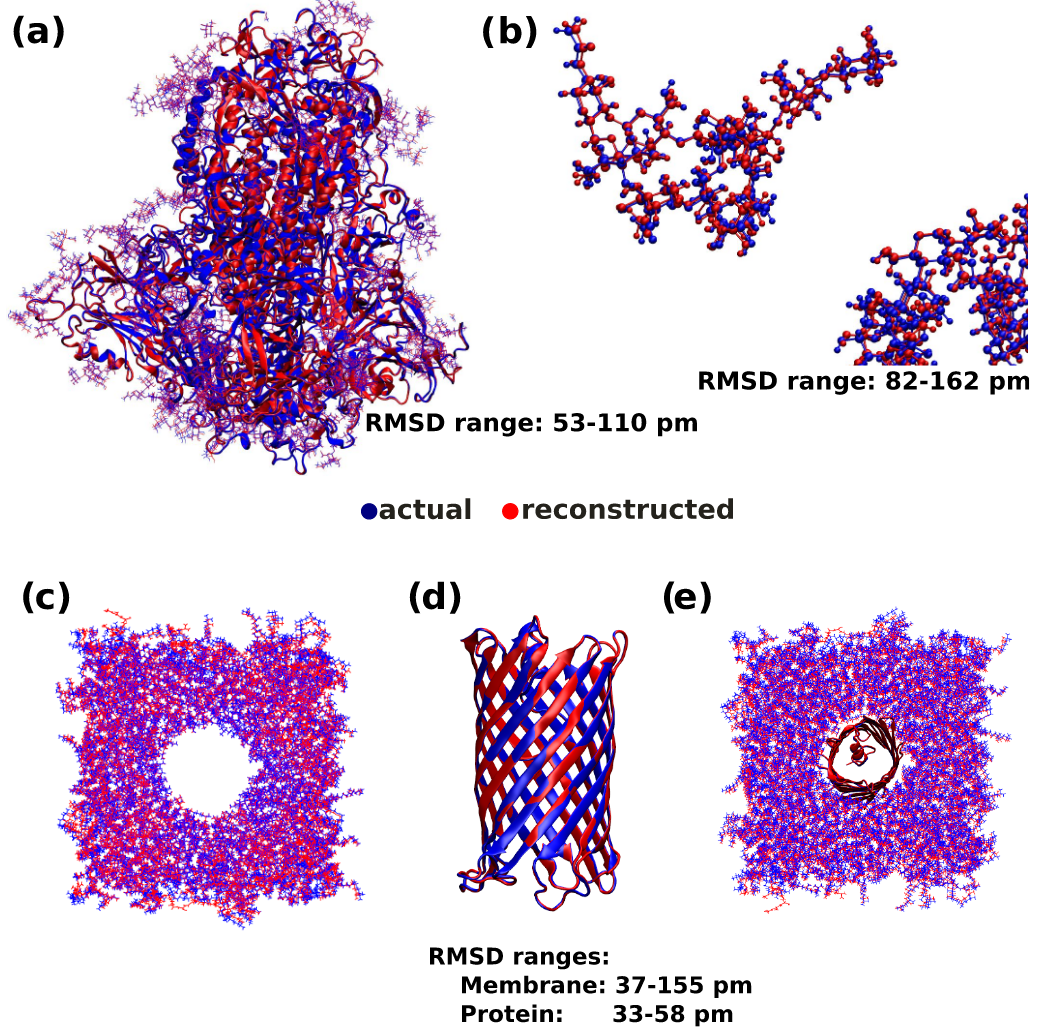
图7 SARS-CoV-2刺突蛋白与膜结合CymA的原始与重建帧对比
进一步测试了细菌膜结合转运蛋白CymA的压缩效果。根据图7结果得出结论,该算法同样适用于膜结合蛋白体系,能够在显著压缩体积的同时保持高保真度的结构重建。
总结
本研究展示了自编码器在高效压缩分子动力学模拟轨迹方面的强大能力。通过建立一套包含参考帧选择、轨迹对齐以及坐标归一化(缩放)的流程,实现了对原子坐标的高精度重建。该方法在多种测试体系中均表现出出色的重建准确性。作者表示,基于自编码器的方法在未来还可扩展至其他大规模数据集的压缩,例如冷冻电镜(cryoEM)数据,这些数据目前因体量庞大而在存储和共享方面面临重大挑战。
参考链接:https://doi.org/10.1021/acs.jcim.5c01294
文章改编转载自微信公众:智药邦
原文链接:https://mp.weixin.qq.com/s/090EqHAmK2U4kmfhJuFx0w?scene=1 | 





















