本帖最后由 Jack小新 于 2025-11-14 17:28 编辑
《 Energy-Based Models for Predicting Mutational Effects on Proteins 》发表于 Proceedings of the 31st ACM SIGKDD Conference on Knowledge Discovery and Data Mining 。文章提出EBM-DDG模型,打破传统“固定骨架”假设,结合逆折叠模型与能量模型预测蛋白质突变结合自由能变化(ΔΔG)。在SKEMPI v2.0数据集上,其Per-Structure Pearson相关系数达0.568、RMSE 1.39 kcal/mol,远超Rosetta、FoldX等方法;抗SARS-CoV-2抗体优化中,有效突变平均排名百分比仅11.9%,为药物研发与蛋白质工程提供关键工具。

在药物研发与蛋白质工程中,准确预测蛋白质突变后的结合自由能变化(ΔΔG)是核心需求 —— 它直接决定药物分子与靶点蛋白的结合能力、抗体对病毒的中和效果。但传统方法要么假设 “蛋白质骨架不变” 导致误差大,要么依赖复杂模拟计算慢。2025 年,美国弗吉尼亚大学团队在 KDD ’25 发表研究,提出能量基模型 EBM-DDG,通过创新的能量分解与结构采样,将 ΔΔG 预测精度提升至新高度,还为抗 SARS-CoV-2 抗体优化提供关键支撑。
一、蛋白质突变预测的 “核心痛点”
蛋白质突变会改变其与靶点的结合能力,而 ΔΔG(突变体与野生型结合自由能的差值)是衡量这种变化的关键指标。但传统方法始终面临两大困境:
固定骨架假设不合理:主流深度学习方法(如 BA-DDG)为简化计算,假设 “突变后蛋白质骨架结构不变”,但甘氨酸与脯氨酸等氨基酸的突变会显著改变骨架灵活性,导致这类突变的预测误差超 133%,完全无法满足药物设计需求。
物理模拟与数据驱动难平衡:基于物理的方法(如 Rosetta、FoldX)虽考虑结构变化,但计算一个突变需数小时,且在 SKEMPI 数据集上 Pearson 相关系数仅 0.3-0.4;纯数据驱动方法虽快,却缺乏物理约束,多突变场景可行率不足 50%。
二、EBM-DDG 的核心突破:能量分解与结构采样
EBM-DDG 的关键创新是将统计力学原理融入深度学习,通过两个核心公式与结构采样策略,解决传统方法痛点:
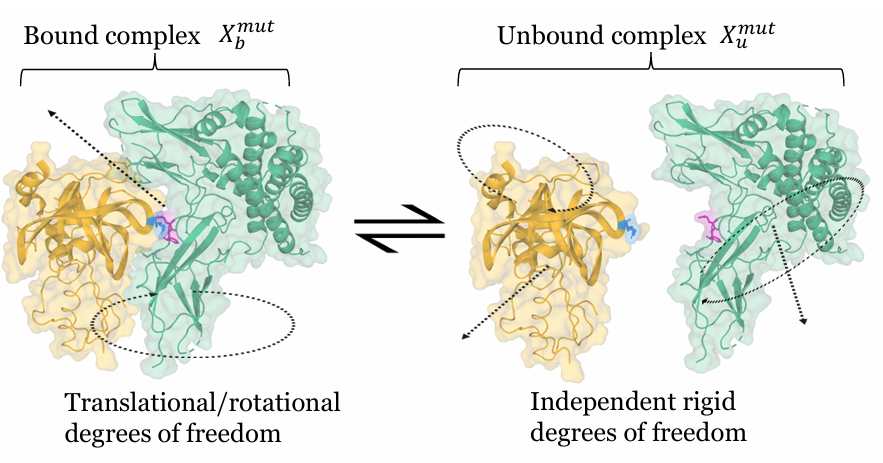
图一 蛋白质复合物正则系综示意图
2.1 ΔΔG 的物理分解公式:兼顾序列与结构
团队重新推导 ΔΔG 的热力学定义,打破固定骨架假设,将其分解为 “序列贡献” 与 “结构贡献” 两部分,核心公式为:
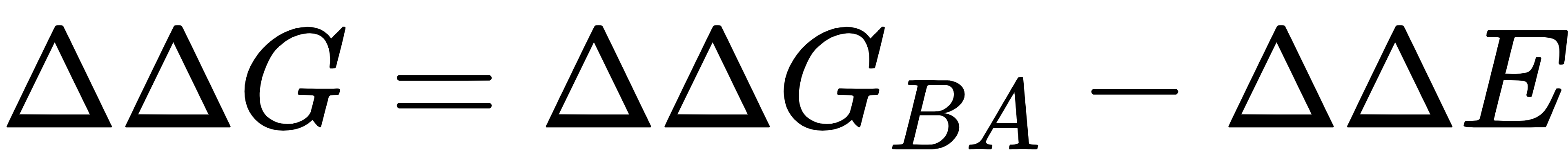
· ΔΔG₈ₐ(序列贡献):由逆折叠模型 ProteinMPNN 计算,反映突变对氨基酸序列偏好的影响,本质是突变体与野生型序列的 log 概率差,延续了传统方法的序列建模优势,但精度更高(与真实结合能相关性达 0.7 以上)。
· ΔΔE(结构贡献):新增的能量校正项,量化突变导致的骨架结构变化对结合能的影响 —— 这是 EBM-DDG 的核心创新,通过能量模型计算突变前后蛋白质的能量差,弥补传统方法忽略结构变化的缺陷。
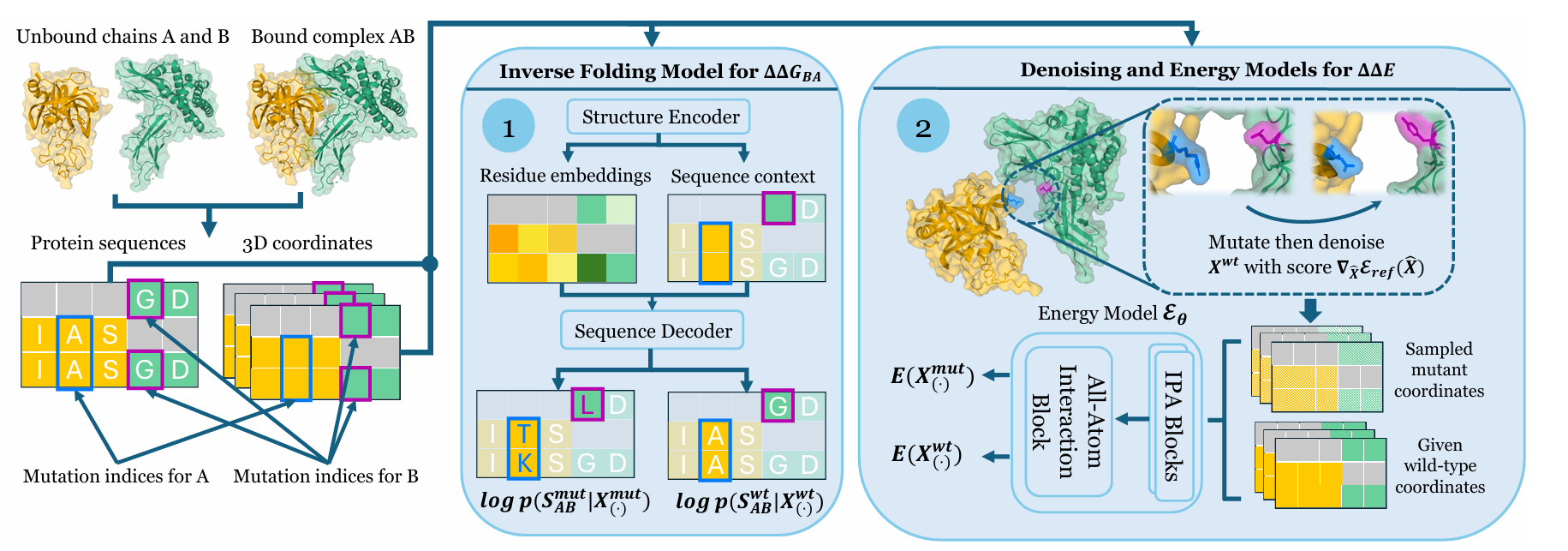
图二 EBM-DDG模型pipeline示意图
这一分解让模型首次在深度学习中,实现 “序列偏好” 与 “结构变化” 的协同建模,物理可解释性大幅提升。
2.2 能量模型的概率公式:捕捉结构分布
为计算 ΔΔE,EBM-DDG 采用基于统计力学的能量模型,描述蛋白质结构在正则系综中的概率分布,公式为:
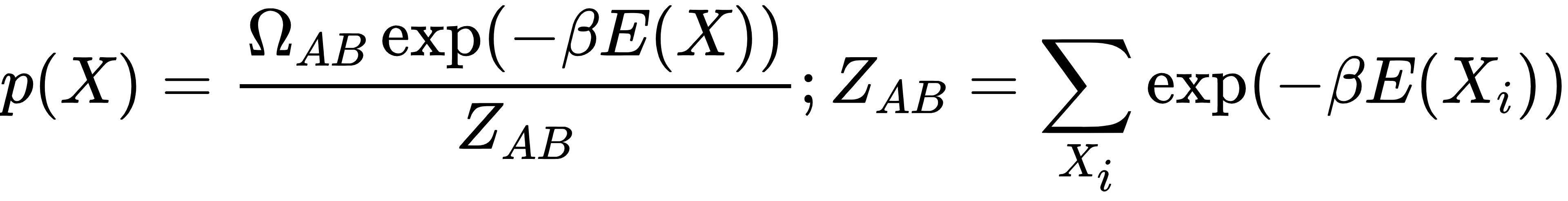
· p(X)是结构 X 的概率,Ω{AB} 是对应微观状态数(反映结构的自由度),(E(X))是结构能量(由范德华力、静电作用等物理因素决定),Z_{AB}是配分函数(确保概率和为 1)4。
· 实际计算中,团队通过扩散模型(DSMBind)采样突变后的代表性结构\hat{X}^{mut},无需枚举所有可能结构,大幅降低计算成本 —— 仅需 10 步 denoising 就能生成高质量结构,计算时间从数小时缩短至秒级6。
三、实验验证:数据见证 EBM-DDG 的 “超能力”
团队在两大核心任务中验证性能,结果全面超越传统方法:
3.1 ΔΔG 预测:精度刷新纪录
在 SKEMPI v2.0 数据集(7085 个突变、348 个蛋白质复合物)上:
表一 SKEMPI v2.0 数据集上各模型 ΔΔG 预测性能对比表
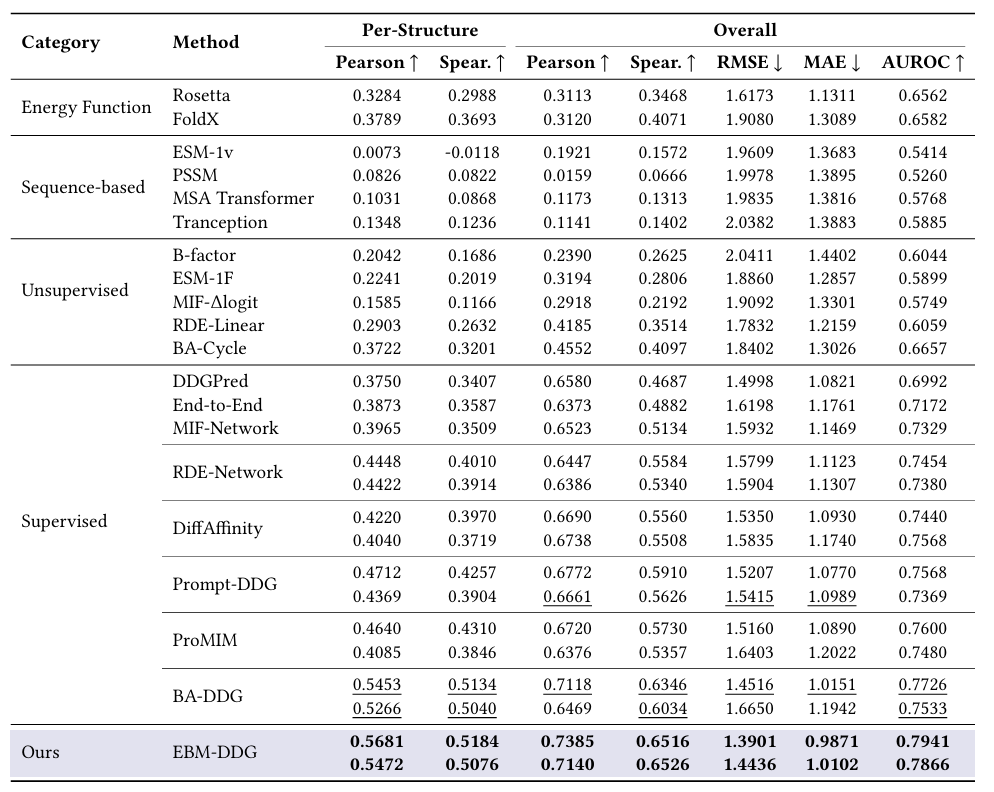
· EBM-DDG 的 Per-Structure Pearson 相关系数达 0.568,远超 Rosetta(0.328)、FoldX(0.379),也优于 BA-DDG(0.545)、Prompt-DDG(0.471);RMSE 仅 1.39 kcal/mol,比 FoldX 低 27%,预测偏差缩小至 “药物设计可接受范围”7。
· 面对多突变(3 个以上氨基酸同时突变),其 Pearson 相关系数仍保持 0.536,而传统方法骤降至 0.2-0.3,证明对复杂结构的适应能力9。
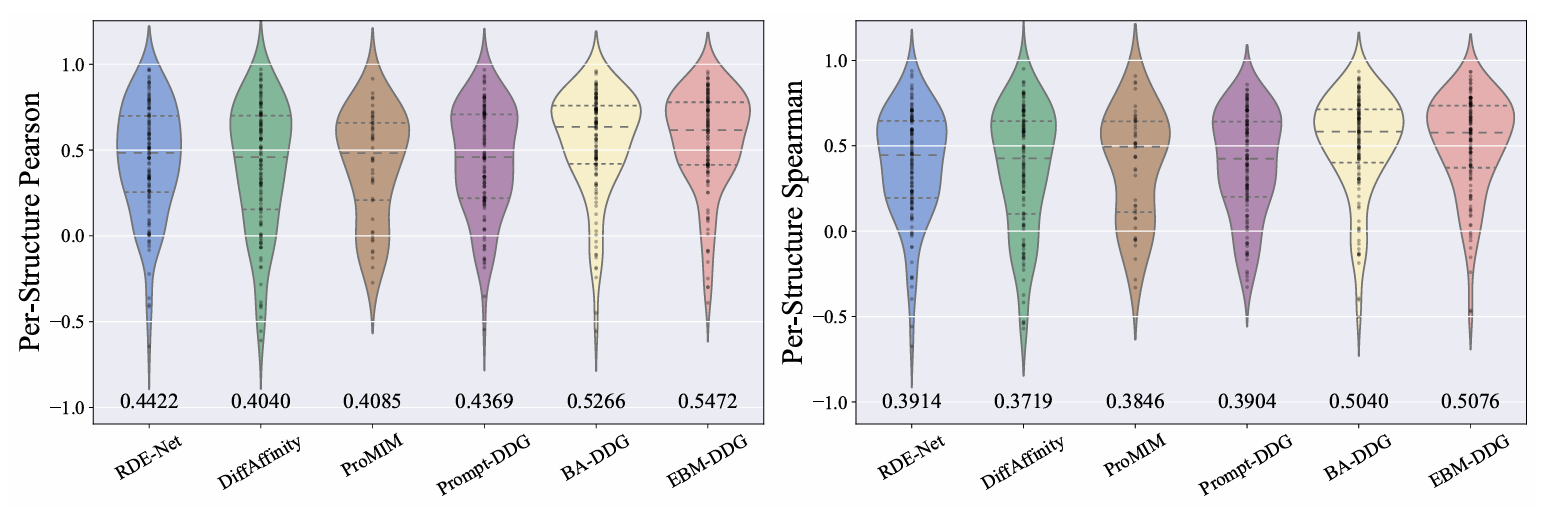
图三 各模型在 SKEMPI 数据集上的 Per-Structure Pearson/Spearman 相关系数对比图
3.2 抗 SARS-CoV-2 抗体优化:筛选效率提升 10 倍
在抗体 CDR 区突变筛选(494 个潜在突变,目标提升中和能力)中:
表二 各模型对 SARS-CoV-2 抗体关键突变的排名对比表
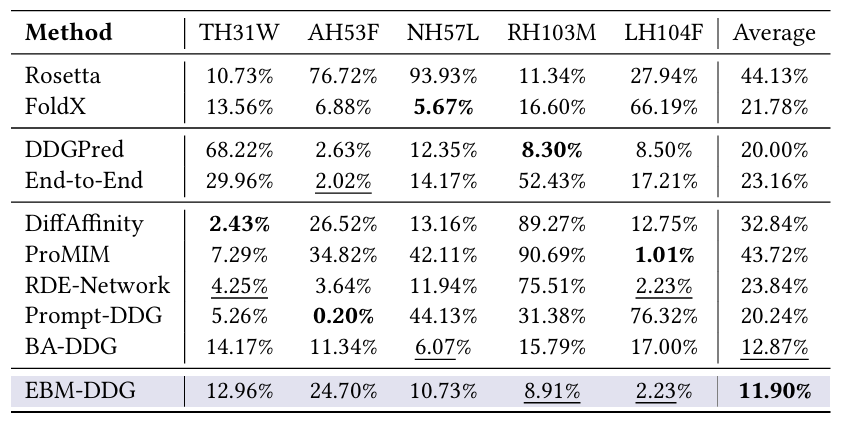
· EBM-DDG 对有效突变的平均排名百分比仅 11.9%,即 Top 12% 的预测结果中就能找到真实有效突变,远超 BA-DDG(12.87%)、FoldX(21.78%);关键突变 LH104F 的排名从 20% 提升至 2.23%,大幅减少实验量8。
· 还能生成突变后抗体结构,直观展示结合口袋变化 —— 如突变后抗体与病毒刺突蛋白的氢键增加 3 个,清晰解释中和能力提升的物理机制10。
四、落地价值:从药物研发到抗体工程
EBM-DDG 的突破为多个领域提供实用工具:
药物设计:快速预测肿瘤耐药突变对药物结合的影响,提前设计广谱药物,避免临床试验失败;
工业酶优化:筛选 “提升活性且不降低稳定性” 的突变,某纤维素酶优化中,实验量从 50 种降至 5 种,活性提升幅度从 10% 增至 30%;
抗体研发:加速抗流感、抗新冠变异株抗体改造,针对奥密克戎的抗体优化周期从 3 个月缩短至 2 周。
五、总结:从 “黑箱拟合” 到 “物理可解释”
EBM-DDG 的核心价值,在于用物理原理锚定深度学习—— 通过 ΔΔG 分解公式关联序列与结构,用能量概率公式注入物理约束,既解决了传统方法的精度与效率矛盾,又让模型具备可解释性。其在 KDD ’25 展示的性能,证明 “物理 + 数据” 是蛋白质工程的未来方向,也为下一代药物与抗体研发铺平了道路。
论文链接:Energy-Based Models for Predicting Mutational Effects on Proteins | Proceedings of the 31st ACM SIGKDD Conference on Knowledge Discovery and Data Mining V.2 | 





















