本帖最后由 Dorian 于 2025-12-25 21:49 编辑
伴随着算法、算力和数据的融合发展, 近年来人工智能(AI)取得了突破性的进展。得益于其在化学和生命科学领域的先驱性应用探索, AI正在成为化学生物学研究的强有力工具, 并展现出整合学科和技术、改变化学生物学研究范式的前景和能力。本文系统回顾了近年来AI在生物成像和谱学解析、生物大分子结构与功能预测、药物发现、精准医学和绿色生物制造五个重要领域的应用, 展示了AI对于化学生物学研究的推动作用。最后, 也结合当前AI技术本身存在的不足和化学生物学研究的瓶颈, 讨论了AI赋能化学生物学领域存在的挑战与未来发展方向。

01 引言
人工智能(artificial intelligence, AI)发展已历经60余年, 期间经历了多次起伏, 但是近20年来, 随着摩尔定律特别是黄氏定律(Huang's law, 以英伟达首席执行官黄仁勋名字命名的定律, 其预测GPU将推动AI性能实现逐年翻倍)驱动的算力提高, 互联网和移动互联网的崛起带来的海量数据积累, 以及深度神经网络算法的崛起, AI在计算机视觉、自然语言处理、自动驾驶等多个领域取得了飞速发展, 已经达到了非常成熟的应用水平, 改变着人类的生产和生活。
以深度学习为代表的AI算法之所以能在近年来取得重大突破, 其本质原因是在算力进步和数据累积基础之上, 新一代的深度学习算法对高维函数处理能力的大幅提升, 而AI所表现出的这种强大的数据降维和表达能力不仅是进行自然语言处理和图像识别的关键, 也是科学研究中处理复杂性的强有力工具, 因此AI正在引发科学研究范式的深刻变化。2024年诺贝尔化学奖和诺贝尔物理学奖均聚焦于AI与科学研究的先驱性结合。诺贝尔化学奖, 颁发给了在蛋白质设计与蛋白质结构预测领域做出开创性贡献的David Baker博士、John Jumper博士以及Demis Hassabis博士; 诺贝尔物理学奖, 授予了John J. Hopfield博士与Geoffrey Hinton博士, 以表彰他们在人工神经网络及机器学习核心原理方面的奠基性工作。这些殊荣所代表的不仅是对过去卓越成果的致敬, 更是对未来科学探索前沿的预告. 科学人工智能(artificial intelligence for science, 简称AI4S)现已成为AI的主战场, AI与不同学科交织, 正不断拓展科学发现的边界, 并展现出整合学科和技术、重新定义科学研究途径、助力抵达未知之境的前景和能力。
面对复杂的分子世界, 化学家们在长期科学探索中积累了大量的物质组成、结构、性质和转化等实验数据, 因此化学学科也是在研究中较早引入数据驱动范式的学科。早在20世纪70年代, Corey等就开发了旨在帮助化学家设计复杂有机合成的合理路线的逻辑与启发式合成分析系统 (logic and heuristics applied to synthetic analysis, LHASA)程序, 是最早尝试将逻辑和启发式方法应用于有机合成规划的系统之一。随着算力、算法和化学大数据在最近几十年的飞速进步, 数据驱动的物质合成、逆合成分析取得了巨大的发展, 通过引入更丰富的数据和更有效的AI算法, 现代的合成规划工具进一步提升了合成路径规划的效率和准确性。
AI的引入, 为化学学科的研究方法带来了深刻的变革。传统的化学研究往往依赖于大量的实验和试错, 而AI则能够通过数据驱动的方式, 快速筛选出有潜力的化合物或反应条件, 大大提高了研究效率。近来, 化学家们更是将AI模型和自动化、机器人技术相结合, 从而实现了从设计、实施到测试整个流程都不需要人干预的智能化自动实验系统。其中具有代表性的是中国科学技术大学江俊团队发展的数据智能驱动的机器化学家。该系统可以自主读取大量化学文献获取先验化学知识, 并自主提出科学假设、设计实验方案; 自主完成化学实验全流程; 通过理论计算建立具备实验反馈的理论预测模型, 并通过机器学习模型和贝叶斯优化算法同时分析实验数据, 为下一次迭代提出新的假设, 实现理论与实验数据的交融。
AI驱动的生命科学研究则是当前最受关注、发展最迅猛的领域之一。从20世纪90年代启动的“人类基因组计划”开始, 生命科学领域就出现了从“实验驱动”向“数据驱动”转变的趋势。也正是高通量测序技术的发展和海量序列数据的积累, 为2021年蛋白质结构预测模型AlphaFold2的横空出世奠定了基础。而AlphaFold系列模型的成功则正式开启了AI在生命科学领域广泛应用的新时代。2024年诺贝尔化学奖得主Demis Hassabis曾这样说: “如果说数学是物理的语言, 那么生物可能是AI语言最完美的描述对象”。国外的谷歌、微软、英伟达、Meta等信息技术产业巨头已经纷纷与生物技术企业开展合作, 加速融合布局。美国哈佛大学、斯坦福大学、麻省理工学院等顶尖大学也已与安进、巴斯夫、拜耳、礼来等医药公司开展了深度学习应用方面的合作, 以驱动药物研发和个性化医疗中的新突破。
化学生物学(chemical biology)是化学与生物学、医学、工程等领域交叉融合的前沿学科, 通过化学理论、方法和技术研究生命现象的本质及调控机制。其核心目标是利用或开发化学工具解析生物分子、细胞、组织、活体等的结构/相互作用及功能; 探索生物过程和疾病发生发展的化学基础和调控新策略, 为生物技术、疾病诊疗和药物研发等提供重要支撑。得益于科学家在化学和生命科学领域开展的先驱性AI应用探索, AI正在成为化学生物学研究的重要工具, 赋能化学生物学研究的各个方面。AI不仅能够通过数据驱动的方式提高研究效率, 还能够整合和分析海量的化学和生物学数据, 发现其中的隐藏规律和关联, 为化学生物学研究提供新的视角和思路。这种研究方法的革新, 不仅加速了化学生物学的发展, 还推动了其与相关学科的进一步交叉融合。
02 AI赋能的化学生物学研究进展
本文将从AI赋能的生物成像和谱学解析、AI赋能生物大分子结构与功能预测、AI赋能药物发现、AI赋能精准医学、AI赋能绿色生物制造五个方面回顾AI技术为化学生物学研究带来的变革。最后我们也将讨论AI赋能化学生物学领域存在的挑战与未来发展方向 (图1)。

图1 AI在生物成像和谱学解析、生物大分子结构与功能预测、药物发现、精准医学、绿色生物制造等方面为化学生物学研究带来变革
2.1 AI赋能生物大分子结构与功能预测
生物大分子(如蛋白质、DNA和RNA等)的结构与其功能密切相关。准确预测生物大分子的三维结构对于理解其生物学功能、设计新型药物以及探索疾病机制至关重要。同时, 通过序列和结构数据预测蛋白质的功能, 为揭示新的药物靶点提供支撑。化学生物学不仅为预测提供实验支撑, 而且其技术手段能够在分子水平上对蛋白质进行修饰与调控, 为验证预测结果、解析密码功能机制提供直接的实验证据, 推动生命科学对遗传信息传递和表达的深层认知。近年来, AI技术的快速发展为生物大分子结构与功能预测带来了新的机遇, 显著提升了预测精度和效率。
2.1.1 蛋白质结构和功能预测
蛋白质是生物体内行使功能的主要生物大分子之一, 它们的结构决定了它们如何与其他分子相互作用来实现它们的功能。通过确定蛋白质结构, 科学家可以绘制蓝图, 指导开发更有效的药物。实验上获得蛋白质结构常借助核磁共振(nuclear magnetic resonance, NMR)、X射线衍射(X-ray diffraction, XRD)、冷冻电子显微镜(cryo-electron microscopy, cryo-EM)等技术手段, 其成本高昂, 而且难以高通量获得结构, 从而为下游基于结构的功能预测和设计增加了技术难度。另一方面, 蛋白质分子是由基本的化学结构单元氨基酸聚合而成的复合物, 因此它的主要特性都由这些基本单元的排布序列所决定。由此衍生的最著名的推论之一, 便是蛋白质的三维结构在很大程度上由组成它的氨基酸序列所决定。
Science杂志曾指出, 蛋白质折叠问题是人类在21世纪需要解决的125个科学前沿问题之一。通过蛋白质结构预测破译“第二遗传密码”, 是生物学中心法则尚未揭示的奥妙之一, 也是目前结构生物学面临的一项具有挑战性的重大基础性研究课题。科学界在过去50多年不断地在尝试如何从氨基酸序列出发预测对应的蛋白质的三维结构。但是由于蛋白质的构象空间大且高度复杂, 因此这个问题极具挑战。
经过多年的努力, 由单序列进行结构预测取得了一些进展, 如Facebook团队的ESM1b模型, 但其精度和可拓展性仍较为有限. 直至2021年谷歌DeepMind团队开发了AlphaFold2(AF2)算法, 它可以仅从序列信息出发, 预测出精度可与实验方法相媲美的蛋白质三维空间结构。DeepMind团队也与欧洲生物信息学研究所(EMBL-EBI)合作推出了AlphaFold蛋白质结构数据库, 涵盖了人类蛋白质组近60%氨基酸的结构位置预测, 这一成就被Nature等学术期刊喻为“前所未有的进步”。AF2发布不久之后, 华盛顿大学David Baker团队也发布了RoseTTAFold, 能够以更低的计算资源消耗达到与AF2不相上下的准确度. 世界上多个团队也都提出了自己的解决方案, 包括北京大学-昌平实验室-华为昇思团队的MEGA-Fold、哥伦比亚大学Mohammed AlQuraishi团队的OpenFold以及深势科技的UniFold等。机器学习技术, 特别是以AF2为代表的深度学习技术, 在蛋白质结构领域的里程碑式成就的核心基础就是相应的数据积累。其中最直接的数据库是几十年来积累的蛋白质结构数据库。
然而, 由于结构解析困难, 时至今日, 已知的蛋白结构仍然只停留在十几万的数量级上. 这对于机器学习, 特别是对数据量极其依赖的深度学习模型, 理论上是远不够的. 真正引发了AF2这样的技术变革, 使得蛋白结构预测进入“大数据”时代的实验技术, 是对蛋白质所对应的基因序列的高通量测序。
自然界在进化的过程中, 产生了大量序列相似, 因而结构相似的近亲蛋白。那些稳定的或是具有功能的结构, 都以相对保守的序列形式在进化过程中保留了下来。此外, 那些在三维结构上靠近的氨基酸, 在进化过程中往往会产生很强的协同突变性。而这些被隐藏在序列中的结构秘密, 构成了现代机器学习模型(包括AF2)来预测蛋白三维结构的重要基础。目前测序方法已经实现了高通量化, 已收录的蛋白序列数据也来到了几亿的数量级, 远远超过已知结构的数据, 且仍在快速增长。因此, 针对某一类感兴趣的蛋白, 随着人们对与它相关的蛋白序列的数据越来越丰富, 我们可以期待利用这些序列的信息越来越准确地预测其结构。2024年, 谷歌DeepMind又取得了重大突破, 发布了蛋白质结构预测领域最新AI模型AlphaFold3 (AF3), 它不仅能够预测蛋白质的三维结构, 还能处理蛋白质与核酸、小分子、离子等生物分子的复合物结构. AF3的关键优势之一是其准确模拟共价修饰的能力, 如键合配体、糖基化以及修饰的蛋白质和核酸残基, 这种能力对于理解生物学过程背后复杂的分子机制至关重要, 有助于为疾病通路、基因组学、治疗靶点、蛋白质工程及合成生物学等领域带来新见解。
AlphaFold系列算法通过深度学习整合多序列比对与注意力机制, 突破了传统结构生物学实验的局限, 实现了接近实验精度的蛋白质结构预测, 为复杂生物机制研究提供全局视角。在病毒糖蛋白机制解析中, AlphaFold结合系统发育分析, 揭示了黄病毒科II类融合系统的保守进化起源, 及肝炎病毒属E1E2糖蛋白的独特结构与脊椎动物感染相关性, 为广谱抗病毒药物及疫苗设计奠定了分子基础。核孔复合体(NPC)研究中, AI建模联合冷冻电子断层扫描(cryo-electron tomography, cryo-ET), 构建了7000万Da的动态支架模型, 发现连接核孔蛋白通过空间组织亚复合体扩大中央孔道, 揭示其构象多样性与核质运输调控机制, 展现出AI与原位技术结合解析亚细胞结构的潜力。
蛋白质递送系统开发领域, AlphaFold预测了昆虫致病细菌的发光杆菌属毒力基因簇(photorhabdus virulence cassette, PVC)尾纤维结构, 指导工程改造使其靶向能力重编程, 以近100%的效率递送Cas9、碱基编辑器等功能载荷至人类细胞, 验证了其在基因治疗与癌症治疗中的应用价值, 体现出AI逆向设计蛋白质的工程化能力。DNA复制机制研究中, 通过AlphaFold筛选互作蛋白发现DONSON作为支架蛋白介导脊椎动物CMG解旋酶组装, 其突变导致的复制缺陷在小鼠模型中重现小头畸形侏儒症表型, 将CMG组装缺陷与疾病直接关联, 加速了致病机制解析。此外, AlphaFold还可以指导小分子药物设计与发现。
如前文所述, AF3可以预测蛋白质、DNA、RNA、小分子等在内的几乎所有生物分子结构和相互作用。而且它在结构预测的准确性方面也取得了长足的进步, 对于蛋白质与其他分子类型的相互作用, 与现有预测方法相比, 实现了至少50%的改进, 而对于一些重要的相互作用类别, AF3的预测准确度实现了翻倍。但AF3也存在局限性, 包括偶尔的立体化学侵犯(stereochemical violation), 如手性误差和原子碰撞; 对某些目标高度准确的预测可能需要生成多个预测并对其进行排序, 从而产生额外的计算成本。另外, 实验上通常是在低温下研究蛋白质以确保其稳定性, 然而范安德尔研究所的研究人员最近的研究揭示某些蛋白质对温度非常敏感, 在体温下结构会发生明显变化, 从而影响其与配体的相互作用位置和方式。而使用现有的AI预测模型, 如AlphaFold系列模型, 目前也只是预测静态的蛋白质结构, 因而对于下游应用(如制药)的作用有限。因此, 仍然需要发展能够捕捉和预测生物大分子生理条件下动态结构变化的方法和模型, 融合体内环境实验技术和AI模型, 并进行交互和迭代。也需要发展能进行高通量计算的跨尺度分子模拟技术, 进行高精度的生物大分子模拟。
DeepMind团队着眼于分子模拟, 开发了通用方法GEMS 通过对“自下而上”和“自上而下”分子片段进行训练, 来构建用于大规模分子模拟的准确机器学习力场。微软研究院也提出了AI2BMD方法实现了对各类蛋白质分子量子化学精度的动力学模拟, 比密度泛函理论(DFT)方法模拟速度快多个数量级, 并实现了对各类蛋白质性质更准确的计算评估。
近年来OpenAI打造的ChatGPT的成功, 使人们看到了大语言模型(large language model, LLM)的威力。相比于小模型数据有限、能力有瓶颈、碎片化情况严重, 以及缺乏规模化复制和涌现能力, AI大模型则具备多个场景通用、泛化和规模化复制等诸多优势。当前的LLM在自然语言处理领域取得了显著进展, 但在理解和生成生物序列(如蛋白质)方面仍然不够出色. 因此, 如何利用大语言模型桥接人类语言和蛋白质语言的鸿沟是一个非常重要的问题。
在近期的一些工作中, 研究者开始使用大语言模型对齐蛋白质序列、结构和功能之间的关系, 开发了蛋白质语言模型(protein language model, PLM), 这些模型巧妙地掌握了蛋白质的基础知识, 并能够有效地泛化以解决各种序列-结构-功能推理问题。例如, 在ProtChatGPT工作中, 研究者设计了将序列和结构通过适配器投射到LLM的结构, 结合用户指定的问题生成关于蛋白质的理解。在InstructProtein工作中, 研究者通过知识因果建模生成从微观层面到宏观层面的知识图谱, 并利用大语言模型生成了许多高质量的指令, 在大语言模型上进行微调后, 可以基于蛋白质序列生成功能描述的文本, 以及利用自然语言提示生成符合要求的蛋白质序列。ESM3采用超大生成式语言模型框架, 可以同时对序列、结构和功能进行快速预测和设计。研究人员使用了超过31.5亿条蛋白质序列、2.36亿个蛋白质结构, 以及5.39亿个带有功能注释的蛋白质数据来训练ESM3, 该模型总共有三种不同的规模, 分别为14亿、70亿和980亿参数。实验表明, 随着模型参数规模的增加, ESM3在生成能力和表示学习上的性能有显著提升, 特别是在生成蛋白质结构时, 980亿参数的模型表现出超越现有模型的强大能力。
ProTokens模型实现了蛋白质三维(3D)结构的深度学习离散化, 证明了蛋白质的骨架3D结构可以被有效离散化成类似于氨基酸的符号, 从而借用操作在氨基酸序列上的序列比对等技术, 实现对3D结构的高效比对、压缩甚至可逆表示(图2)。ProTokens模型的核心思想是将蛋白质的连续3D结构转化为离散的“Token”表示。这一思想源于蛋白质物理学中的亚稳态理论。尽管蛋白质结构在笛卡儿坐标空间中可以连续变化, 但其稳定态的数量是可数的。基于这一理论, 研究者提出了概率性Token化理论, 将蛋白质结构的连续分布分解为离散部分(亚稳态)和连续部分(亚稳态内的波动)。这种方法不仅能保留蛋白质结构的关键信息, 还能使蛋白质结构更易于AI模型处理,为蛋白质设计开辟了新的可能性。最近发布的ProtTeX模型也是通过结构Token化和序列Token化将蛋白质问题转化为语言建模任务, 实现了蛋白功能预测、结构生成与分析、多轮链式推理(chain-of-thought)和定向蛋白设计。不过, 需要强调的是, 在蛋白质功能预测研究中, 孤儿蛋白(功能未知蛋白)的预测仍是亟待解决的关键科学难题。这类蛋白因缺乏功能已知的同源蛋白或明确的结构特征, 导致传统的序列比对和结构预测方法效果有限。
为突破这一瓶颈, 研究人员亟需发展基于蛋白质语言模型的新型预测技术, 利用大规模预训练模型深度挖掘蛋白质序列中隐含的功能特征. 同时, 通过将转录组、蛋白互作网络、代谢通路等多组学数据整合到蛋白质语言模型中, 构建多维度的功能关联网络, 则有望显著提高预测可靠性。可以预见,通过融合人工智能模型、多组学数据整合及结构预测等多维度方法, 将有望系统解析未知功能蛋白的生物学机制, 从而填补当前功能注释的空白。未来,当更大规模、更丰富的蛋白质数据能被大语言模型所利用, 蛋白质语言模型就有可能推断出远远超过人类认知极限的蛋白质潜在规律或深层结构, 从而为蛋白质科学研究开辟崭新境界。
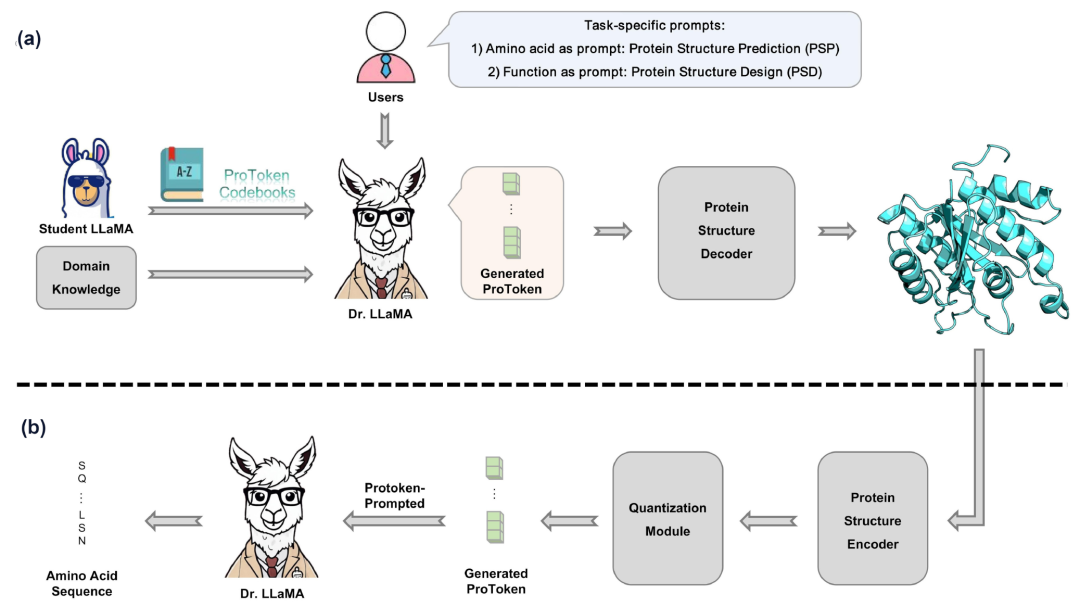
图2 基于大语言模型的ProTokens示意图. (a) 解码器功能: 配备解码器的Dr. LLaMA能够以ProTokens形式生成蛋白质3D结构, 既可根据指定的氨基酸序列提示进行结构预测, 也可根据功能需求提示完成结构设计. (b) 编码器功能: 配备编码器的Dr. LLaMA能对蛋白质主链进行逆向折叠, 推导出对应的氨基酸序列
2.1.2 RNA结构预测
RNA的序列比对同样可以被应用于其三维结构及结构信息预测,但目前该领域还主要集中在二级结构预测方面,其三维结构预测较为困难和耗时。该领域的数据集规模相对较小,PDB单链RNA数目和代表性的RNA三维结构数据集包含的结构单元数均为万量级。对RNA结构预测,深度学习数据增强方法可能帮助生成更多训练数据,从而帮助构建更准确的预测模型; 也可以发展高通量分子模拟方法以帮助三维结构预测以及进行数据生成; 还可能通过实验-计算结合的方式,用相对少量实验数据帮助结构建模。
2021年斯坦福大学Ron O Dror团队用几何深度学习开发的ARES系统,仅需18个已知RNA结构作为训练数据,即可突破传统深度学习的数据依赖局限,实现RNA结构的精准预测。清华大学和深圳湾实验室团队在RNA结构预测的数据库和模型建设中具有较好的基础,最近香港中文大学、复旦大学等机构的研究人员则开发了一种名为RhoFold+的深度学习方法,用于从头预测RNA 3D结构。该方法基于RNA语言模型,并在约2370万个RNA序列上进行了预训练,解决了数据稀缺性的问题。RhoFold+提供了完全自动化的端到端流程,在单链RNA建模方面表现出很高的准确性,并具有出色的泛化能力,能够捕捉螺旋间夹角和二级结构等局部特征。在RNA-Puzzles和CASP15天然RNA靶标的评估中,RhoFold+的表现优于现有方法。尽管RhoFold+取得了显著的成果,它仍然存在一些局限性,如依赖多序列比对(multiple sequence alignment,MSA)、难以预测大型复杂RNA结构以及难以模拟RNA的动态特性和与其他分子的相互作用。未来的研究方向包括整合探测数据、分子动力学和能量函数等方法,以提高RhoFold+的准确性,并增强MSA提取过程和RNA相互作用预测能力。
AI在RNA结构与功能预测研究领域实现了多维度突破,构建起从基础研究到临床转化的完整技术链条,促进了RNA化学生物学研究。例如,美国国家癌症研究所王运星团队针对柔性RNA结构解析难题开发了HORNET方法,在生理条件下实现了单分子RNA动态构象的可视化解析。该技术成功捕获HIV-1 Rev响应元件RNA (RRE RNA)的五种异质构象,揭示了其构象异质性直接影响Rev蛋白结合效率,并设计出结合力超越天然蛋白3倍的多肽分子,在小鼠模型中降低病毒载量90%。又如,加州大学Gene W Yeo团队构建的HydRA系统,实现了超万级RNA结合蛋白(RBP)的精准预测。他们进一步结合实验发现了数百个新型RNA结合结构域,并证实其功能活性,极大拓展了RNA调控网络的认知边界。
2.1.3 生物大分子相互作用
DNA、RNA、蛋白质作为中心法则的三种重要分子,细胞生命过程的实现依赖于DNA、RNA、蛋白质等分子之间的复杂相互作用,目前人们对其中具体的作用形式已经有了较为丰富的认识。蛋白质是多数细胞活动的直接执行者,其功能实现往往需要分子间相互作用,包括但不限于蛋白-蛋白、蛋白-RNA、蛋白-DNA等类型; DNA-RNA相互作用与转录和转录调控直接相关; DNA间的远程物理相互作用与其调控往往需要转录因子蛋白辅助; RNA直接参与蛋白翻译,还可以与DNA、蛋白质一同通过液液相分离的机制形成细胞内的无膜细胞器,调控基因的转录与翻译。理解这些二体乃至多体相互作用不仅有助于增强对生命过程中调控关系的理解,更有助于对这些调控关系进行干预,从而预防或治疗疾病。
基于生物计算预测蛋白间相互作用及其变化,可以促进抗体药物等蛋白药物设计以及生物制药的发展,是研究的难点之一。以DeepMind开发的AlphaFold-Multimer为代表的蛋白间相互作用的深度学习预测模型是近期这个方向上的重要进展,但由于构成复合物的多条子链之间往往缺少共进化信息和全局模板信息,所以目前的深度学习预测模型大多在多链复合物的结构预测中表现并不理想。针对这一问题,Feng等提出了一个用于蛋白复合物构象预测的通用框架——ColabDock,它是一个由稀疏实验约束引导的蛋白质-蛋白质对接结构预测通用框架。通过在构象搜索过程中使用梯度反向传播替代传统蛋白对接软件中的快速傅里叶变换,该方法有效整合了蛋白结构预测深度学习模型的能量景观和稀疏实验约束,可以自动搜索满足两者的构象,同时也能容忍约束中的冲突或模糊性。另外,ColabDock可以利用不同形式和来源的实验约束,而无需进一步进行大规模重新训练或微调。测试显示,ColabDock不仅在具有模拟残基和表面约束的复杂结构预测中优于HADDOCK和ClusPro,而且在结合核磁共振化学位移扰动和共价标记辅助的情况下也表现出色。
北京大学/昌平实验室高毅勤团队也发展了可以整合多种实验信息辅助蛋白复合物结构预测的原创性方法和模型GRASP(广义约束辅助结构预测模型,generalized restraints assisted structure predictor),通过在AI模型中引入实验约束和分子模拟采样,把有限制的结构生成和强化学习结合起来。该方法可以应用于XL-MS、NMR、共价标记(covalent labeling,CL)、深度突变扫描(deep mutational scanning,DMS)、化学位移微扰(chemical shift perturbation,CSP)、氢氘交换质谱(hydrogen-deuterium exchange mass spectrometry,HDX-MS)等多种类型的实验约束整合,并进行抗原-抗体等蛋白质复合物结构预测,预测精度上超越了AlphaFold-Multimer和AlphaFold3。更为重要的是,该方法能够高效利用稀疏的实验信息实现蛋白相互作用组的高通量建模,实现亚细胞层级的蛋白相互作用组搭建和体内动态相互作用建模,为进一步的疫苗和抗体研发、疾病诊断、靶点发现和药物设计等提供基础。
对比蛋白-蛋白相互作用预测,关于DNA-蛋白、DNA-DNA和DNA-RNA的相互作用研究,目前更是困难重重。这些问题的解决,需要整合多尺度、多组学信息,分类构建一系列分析技术与方法。在同种分子层面上,整合多种一维组学数据以对DNA层面的三维相互作用进行预测; 进一步开发蛋白间相互作用的预测模型与结合蛋白药物的设计。发展和整合基因组、表观遗传组、蛋白组等多组学数据集,开发多模态方法,以适应生物体系的多层级特性,可以增进对生物大分子调控关系的理解。这些方面的综合性研究,特别是将三维基因组学、表观遗传学和蛋白组学结合起来研究多种大分子的相互作用的工作处于起步阶段,将可能带来重要的研究范式变革。例如,北京大学高毅勤团队通过分析染色质三维结构特征发现,染色质结构包含的基因邻近信息可能参与指导转录与翻译层面的分子互作关系,进而构建基因调控网络。首先,从DNA到RNA层面,结直肠组织染色质三维结构中基因的邻近关系与基因的共表达存在对应关系。这说明正常组织染色质三维结构的长程相互作用在基因转录调控中可能起到了重要作用,使得序列距离较远的基因也能够共享相似的转录环境,包括转录因子和表观遗传信号,从而实现共调控与共转录。其次,从DNA到蛋白质层面上,他们发现邻近基因翻译出的蛋白也更倾向于具有物理相互作用。这些发现拓展了对中心法则的理解,即除了一维序列信息的传递,基因间的调控关系也可以储存在染色质三维结构中(DNA层面),通过基因的转录共调控(RNA层面)从而实现调控下游蛋白间的相互作用(蛋白层面)。
2.2 AI赋能的生物成像和谱学解析
生物成像和生物谱学解析作为化学生物学研究中的两大核心技术, 在解析生命分子机制、动态过程及疾病机理中发挥着不可替代的作用。AI通过自动化解析、多模态整合及多尺度分子模拟, 正在重塑生物成像与谱学解析的研究边界。
2.2.1 生物成像
生物成像技术通过高时空分辨率的成像手段,实现对生物分子活性和细胞过程的实时动态观测,其为化学生物学研究提供了强有力的工具和方法。自20世纪50年代至今已有多项诺贝尔奖与显微成像技术相关,之后成像技术发展迅猛,新技术层出不穷。2014年诺贝尔化学奖被授予研制出超分辨率荧光显微镜的三位科学家,他们将荧光显微成像的分辨率带入到“纳米时代”,极大地推动了生命科学领域的研究工作。然而,超分辨显微成像在数据采集、重建和分析中仍面临噪声干扰、成像速度限制、动态过程捕捉困难等挑战。近年来,得益于AI技术的快速发展,深度学习被用于克服超分辨显微技术的各种缺陷。
单分子定位显微镜(single-molecule localization microscopy,SMLM)通过随机激发荧光分子并定位重建来实现超分辨成像,但其存在时间分辨率低、光毒性、光漂白、分子定位精度和速度低等问题。为了提高重建速度,Nehme等提出了深度学习驱动随机光学重建显微法(deep stochastic optical reconstruction microscopy,Deep-STORM),该技术利用卷积神经网络(convolutional neural networks,CNN)从SMLM的稀疏数据中重建超分辨图像,显著提升了定位精度和信噪比。接着,Li等受Deep-STORM启发,进一步结合递归神经网络提出了深度递归监督网络(deep recurrent-supervised network)-STORM (DRSN-STORM); Speiser等则提出了一种基于U-Net网络的深度上下文相关(deep context dependent,DECODE)架构,用以在单分子定位中区分真实信号与随机噪声; 也有研究者基于神经网络开发了ANN-PALM、DBlink等方法来减少图像重建所需帧数,这些方法都提高了成像速度和定位精度。受激发射损耗显微术(stimulated emission depletion microscopy,STED)是主流的超分辨技术之一,它通过高斯激发光和环形光束的配合,实现超分辨成像。STED实现超分辨的关键在于损耗光的功率以及受激辐射与自发荧光相互竞争中的非线性效应,淬灭光功率越强,空间分辨率越高,但使用强耗损光的同时会带来光漂白、光毒性、光损伤等问题。此外,对于厚样品STED的轴向分辨率仍有待提升。为了提高成像速度,减少光损伤,Ebrahimi等借助U-Net和残差通道注意力网络(residual channel attention network,RCAN)架构提出了多阶段渐进图像恢复(multi-stage progressive image restoration,MPRNet)的方法,能够使STED的像素停留时间减小1~2个数量级,极大提升了成像速度,进而减少了对样品的光漂白与光损伤。
此外,还有研究通过深度对抗网络(deep adversarial networks,DAN)、结合单螺旋点扩散函数与深度学习算法,进一步提升了STED的横向和轴向分辨率。研究者也尝试使用深度学习将非超分辨成像技术所成图像直接转换为超分辨图像。例如,Wang等使用生成对抗网络(generative adversarial networks,GAN)实现了共聚焦图像与STED相匹配的分辨率; Huang等提出的双通道注意力网络(two-channel attention network,TCAN)提高了图像分辨率等。AI技术正逐步重塑超分辨显微成像的全流程: 从数据采集(实时去噪、自适应光学)、重建(分辨率突破)到分析(动态追踪、功能解析)。未来,随着物理驱动AI模型、边缘计算与跨学科方法的进一步发展,超分辨成像将迈向更高维度(4D时空成像)、更高通量(全组织尺度)及更高智能化(自主实验设计)。这一技术革新不仅推动基础科学研究,也为精准医学诊断(如病理切片超分辨分析)和新药研发(如单分子药物靶点追踪)提供了强大工具。
生物大分子的结构解析对于理解其功能和相互作用至关重要。通过揭示生物大分子的三维结构,研究人员能够更深入地了解其如何参与生命过程,如酶催化、信号传导和基因表达等。cryo-EM是近年来在结构生物学领域最重要的生物成像技术,被科学家称为“诺奖助手”。然而,传统的cryo-EM单颗粒分析重构方法往往仅生成一个静态的三维结构,无法进行动态构象分析。此外,生物大分子之所以能实现众多关键的生物学功能,很大程度上得益于其卓越的柔性结构特质。然而,正是这一柔性结构特质,成为了研究人员对其进行高精度结构解析的主要障碍。因此,结构生物学领域的一个重要挑战就是如何高分辨率地解析生物大分子的三维结构,尤其是其柔性区域结构,并通过重建其动态过程来理解其生物学功能。
AI的引入为cryo-EM技术的发展带来了新可能,冷冻电子显微镜数据处理的关键环节,包括粒子选取、三维重建、分辨率确定、图像锐化和模型构建等,都可以利用AI来优化和增强。例如,Liu等开发的spIsoNet技术,通过自监督深度学习显著提高了生物大分子重建的质量,增强了对齐精度和角度各向同性。马剑鹏团队发展了冷冻电镜密度图重构算法OPUS-DSD,不但能够成功地解析因传统解析方法无法分辨而缺损的生物大分子(如蛋白质、核酸或蛋白质/核酸复合物等)结构,并且能高效、精准地分辨出柔性结构域在受测样品中的构象分布。他们也开发了蛋白质侧链建模技术OPUS-Rota5,经过OPUS-Rota5侧链修正后的结构具有更高的分子对接成功率。cryo-EM技术的持续发展使研究人员能够研究更复杂、更具挑战性的超大生物大分子机器的结构和功能。例如,孙飞团队全面介绍了利用冷冻电子显微镜技术对核孔复合体的研究,特别强调了通过结合最新冷冻电子显微镜技术和AI建模技术实现亚纳米分辨率的突破性进展。颜宁团队则提出了一个名为CryoSeek的新策略,将冷冻电子显微镜作为一种观察工具,结合AI辅助的自动建模和生物信息学分析,发现了自然界中完全未知的新型生物实体。在动态过程捕捉上,具有代表性的工作是2022年北京大学毛有东团队将AI应用于提升时间分辨冷冻电子显微镜的分析精度,解析了蛋白酶体降解底物的13种中间态构象,揭示了USP14调控的动力学机制。
结构生物学研究的未来目标是在细胞环境中进行原位结构研究,cryo-ET技术使这一目标成为现实,开创了结构生物学的新时代。与单颗粒分析不同,cryo-ET能够直接对细胞切片进行成像,并通过倾斜系列图像重建出切片的三维结构,从而揭示生物大分子在其天然状态下的空间组织和相互作用。然而,传统的cryo-ET技术同样面临诸多挑战。例如,由于电子束的辐射损伤,cryo-ET通常需要使用极低剂量的电子束采集数据,这会导致信噪比较低。同时,倾斜样品平台也会导致成像的对比转移函数出现空间变化,进一步限制成像的分辨率。此外,cryo-ET的数据中保留细胞内各种分子,这为从其中辨别分析特定分子带来了巨大挑战。更重要的是,生物大分子在细胞内的动态行为和构象变化往往被“冻结”在某一时刻,现有的cryo-ET数据分析方法也是基于静态假设,所以难以捕捉动态过程的细节。而结合机器学习和先进图像处理技术,则有望从cryo-ET数据中还原更加精细的分子结构和动态特征,从而深入探索生物大分子在复杂生理环境中的功能机制,以及不同大分子的协作。
AI与冷冻电镜的协同创新,不仅解决了传统结构生物学的效率瓶颈,更开启了动态结构与复杂体系研究的新纪元。未来,随着算法提升、多模态数据整合和自动化平台的普及,这一技术组合将在基础科学和医学中释放更大潜力。
2.2.2 生物谱学解析
生物谱学技术(如质谱、核磁共振)通过高灵敏度的分子检测,提供生物分子的定性与定量信息,被广泛应用于化学生物学研究中:质谱(mass spectrometry,MS)技术可鉴定蛋白质结构、分析磷酸化/糖基化修饰,并实现蛋白质组定量;质谱结合色谱分离技术,被用于解析代谢物谱以揭示疾病标志物;通过设计小分子探针干扰特定信号通路,并结合质谱分析探针-靶标结合位点,可以用于研究细胞坏死或自噬的调控机制。
谱学方法的应用一直受制于谱学数据的解析效率和难度,传统的人工或半自动化解析方法不仅效率低下,而且容易引入主观偏差,严重依赖于实验者的经验。深度学习模型的应用则可以极大地减少人为错误,提高数据处理的速度和准确性。例如,清华大学陈春来团队发展的DEBRIS方法,通过精准识别单分子荧光轨迹的局部特征,并允许根据实验设计灵活调整分类标准,实现了在不修改神经网络结构的前提下,对双色/单色实验条件下的稳定和动态单分子荧光信号进行准确识别。中国科学院大学和温州医科大学的研究团队,则通过使用拉曼光谱结合卷积神经网络研究人体肝组织样本,以快速、非破坏性和无标签的方式将癌组织与邻近的非肿瘤组织区分开来。
NMR方法是一种以原子分辨率解析更贴近蛋白质在实际环境下的溶液态构象与动态结构的方法,然而该方法存在数据解析速度慢的问题,平均单条蛋白需要领域专家投入至少数月,而其中大部分时间都消耗在实验数据的解析和归属上。高毅勤团队发展了AI+约束结构预测模型RASP,并在其基础上开发了核磁共振增强光谱(nuclear overhauser enhancement spectroscopy,NOESY)自动解析方法——蛋白折叠结构辅助的共振峰指认(folding assisted peak assignment,FAAST),实现了NMR数据解析时间从数月到数小时的缩短。陈忠团队将物理信息嵌入仿真数据驱动的神经网络模型中,提出基于深度学习的多维拉普拉斯磁共振快速重建算法DLEMLR,克服了拉普拉斯反演的病态性及提高重建谱图的分辨率,并将重建时间缩短至秒级。基于质谱的蛋白质组学是蛋白质鉴定的核心技术,但其数据处理面临高噪声、高维度等挑战。
AI在质谱分析中不仅可以加速数据处理和解读,还通过预测模型和自动化技术革新了实验设计和结构解析,成为推动蛋白质组学发展的核心驱动力。通过深度学习算法可以显著提升数据独立采集(data independent acquisition,DIA)的复杂谱图解析能力,使肽段识别数量翻倍,减少假阳性。利用深度学习生成预测谱库,辅助DIA数据分析,也可以提高低丰度肽段的检测灵敏度。AI也能用于预测肽段的洗脱时间、离子化效率及“proteotypic”肽段(易检测的代表性肽段),优化实验设计。在交联质谱(cross-linking mass spectrometry,XL-MS)中,通过整合AlphaFold2等AI工具,解析蛋白质相互作用网络和结构模型,则可以提升交联数据的结构背景解释。
2.3 AI赋能药物发现
化学生物学研究的核心在于利用化学工具揭示生命过程的分子机制,并直接干预这些机制以解决相关问题,因此药物发现是化学生物学的终极目标之一。然而, 传统药物研发面临“双十”魔咒,即新药研发通常需要花费10年时间、10亿美元。如何打破这一魔咒,AI被寄予了厚望。由于在数据降维、模式识别及生成能力上的优势,AI正在重塑药物研发全流程,有望将靶点发现、虚拟筛选、药物分子从头设计等环节的效率大大提升(图3)。
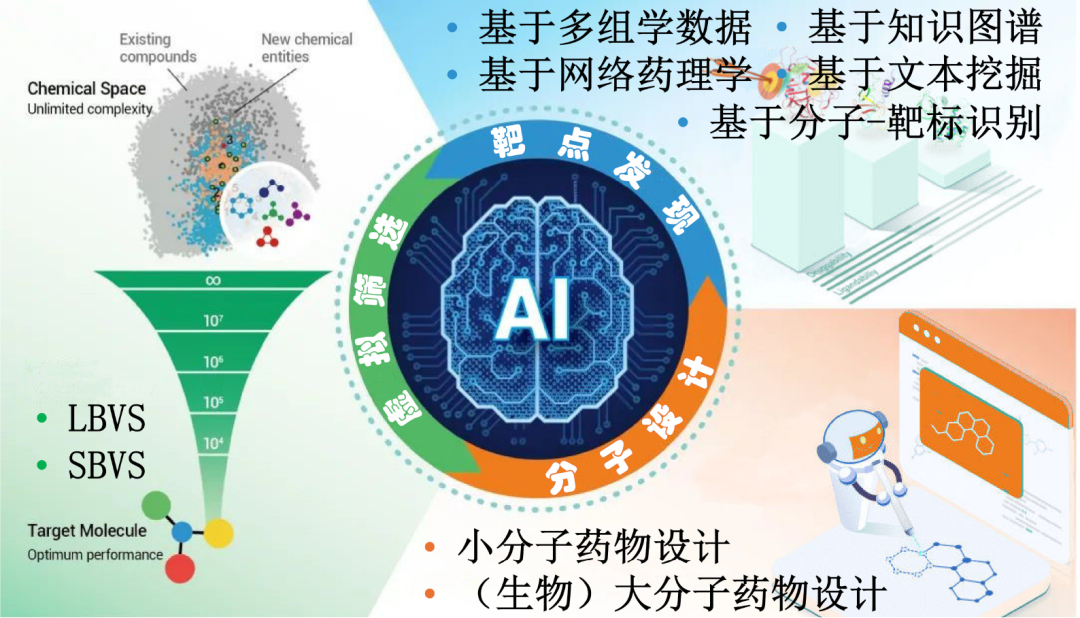
图3 AI正在重塑药物发现中的靶点发现、虚拟筛选和药物分子从头设计等关键步骤
2.3.1 靶点发现
在新药研发的整个链条中,一个新靶点的发现往往会带动一批新药产生,推动临床治疗的突破。传统的药物靶点发现方法主要依赖于生物学实验、化学筛选和生物信息学分析,旨在通过系统性手段揭示疾病相关分子机制并筛选潜在药物作用靶点。由于AI擅长分析海量复杂数据集,在其中挖掘隐藏模式,因而近年来AI技术正成为发现新靶点的利器。
(1)基于多组学数据的靶标发现。随着高通量测序技术的进步,海量的组学数据(如基因组学、转录组学、蛋白质组学、代谢组学等)不断产生。多组学数据从不同角度为研究人员提供了分子相互关联的信息,通过对这些大规模组学数据进行处理和分析,可以鉴别出在与特定疾病相关的生物过程中扮演重要角色的基因或蛋白质,从而促进药物靶点发现的研究。然而,处理和分析这些复杂且高维组学数据极具挑战性。通过机器学习和深度学习算法来整合多组学数据,则可以从大规模组学数据集中学习潜在知识,高效地识别关键生物标志物和可成药靶点。例如,为了识别肌萎缩侧索硬化(amyotrophic lateral sclerosis,ALS)的治疗靶点,Pun等结合多种基于生物信息学和深度学习的模型,使用疾病特异性多组学和基于文本的数据进行训练,以优先考虑可药物基因,揭示了ALS治疗的18个潜在靶点。Fabris等建立了一种基于深度学习的方法,通过学习从基因或蛋白质特征中检索到的模式来识别与多种年龄相关疾病的人类基因。
(2)基于分子-靶标识别的靶标预测。预测并确证活性分子的靶标是阐明药物作用机理的重要步骤。传统的靶标识别方法主要是同位素示踪法、紫外及荧光光谱法,效率较低。目前常用的方法是基于基因组学和蛋白组学的高通量筛选方法,但仍存在成本较高、实验周期长、不具有普适性等缺点。AI可通过深度学习算法,建立分子-靶标数据库,从而高效预测药物分子的潜在靶标。例如,Nelson等提出的基于CNN和全连接神经网络(fully connected neural network,FCNN)的端到端深度学习模型,无需依赖手工设计的描述符,直接从蛋白质序列(氨基酸序列)和化合物SMILES字符串中提取特征,在药物-靶标相互作用(drug-target interactions,DTI)预测任务中表现优秀。
(3)基于生物医药知识图谱的可成药靶点发现。将知识图谱技术与系统生物学结合构建生物医药知识图谱(biomedical knowledge graphs)已开始在生物医药领域发挥关键作用。通过与特定疾病的背景相结合,交叉检验多源异质的生物医药数据库(蛋白质组数据库、蛋白质相互作用数据库、药物-靶点关系数据库等),生物医药知识图谱可以获取其中的内在关联,加速靶点识别。例如,Zitnik实验室开发的精准医学知识图谱(precision medicine knowledge graph,PrimeKG)项目,整合了20个高质量的生物医学资源,涵盖了17080种疾病、7957种药物,通过知识图谱中的药物-疾病关系可以帮助药物研发人员识别潜在的药物靶点和治疗方案。郑杰课题组与合作者提出了基于知识图谱和图神经网络的模型KG4SL,通过知识图谱来揭示合成致死(synthetic lethality,SL)基因背后的生物学机理,有望加速癌症药物靶点发现。
(4)基于网络药理学的靶标发现。网络药理学的概念由英国药理学家Andrew L。Hopkins于2007年首次提出,其利用生物分子网络分析方法,选取特定节点进行新药设计和靶点分析。网络药理学突破传统的“一个药物一个靶标,一种疾病”理念,代表了现代生物医药研究的哲学理念与研究模式的转变。以系统生物学和网络生物学基本理论为基础的网络药理学具有整体性、系统性的特点,注重网络平衡(或鲁棒性)和网络扰动,强调理解某个单一生物分子(如基因、mRNA或蛋白等)在生物体系中的生物学地位和动力学过程要比理解其具体生物功能更为重要,揭示药物作用的生物学和动力学谱要比揭示其作用的单个靶标或几个“碎片化”靶标更重要,对认识药物和发现药物的理念产生了深远影响。AI非常擅长分析基因、蛋白质和通路的相互作用网络,以确定疾病进展的关键节点。未来网络药理学的研究将会涉及更多的多模态数据,如基因组学、转录组学、蛋白质组学、代谢组学等数据。面对多维度数据,人工智能技术在这方面的应用已经开始受到广泛关注,未来的网络药理学也将借助这些技术的发展,实现更加智能化和高效的分析和预测。
(5)基于生物医药文本挖掘的靶点发现。人们在生物学基础研究和临床研究中积累了大量数据,但这些数据“互不关联”地分散存储在海量的研究文献中,它们之间的潜在关联难以被人类发现。大语言模型LLM具备理解自然语言和解析复杂科学概念的能力,因而LLM驱动的AI方法具有强大、高效的学习分析能力,能够将散布在大量文献中的关联关系挖掘出来,从而推动新机制、新靶点的发现。微软的BioGPT和英矽智能的ChatPandaGPT (集成于英矽智能的人工智能驱动的靶点发现和生物标志物识别平台PandaOmics)就致力于能够将疾病、基因和生物过程相互关联,从而快速识别疾病发生和发展的生物学机制,并发现潜在的药物靶点和生物标志物。然而,这些模型通常基于人类生成的文本进行训练,可能无法判断输入数据的准确性和适用性。因此,它们可能会无意中延续人类的偏见和先入为主的观念。此外,由于这些模型严重依赖已发表的数据,它们在识别真正新颖靶点方面的潜力可能有限。因此,后续的研究中需要认识到这些局限性,并辅以其他模型的使用,以确保发现真正新颖且相关的靶点。
2.3.2 虚拟筛选
确定了靶点后,药物研发的后续任务基本上就是寻找一个具有临床功效的先导化合物。但是,由于在化学文摘数据库中已注册的化合物数量超过7000万个,再加上其他可能存在的无穷无尽的化合物,可以成为候选药物的化合物数量难以统计,因此如何在这么巨大的化学空间中进行搜索是一个高难度的问题。目前,已有许多工具和方法来帮助我们发现先导化合物,它们通常可以被分为两类: 高通量筛选(high throughput screening,HTS)和虚拟高通量筛选(virtual high throughput screening,vHTS)。尽管实验性高通量筛选能够考虑生物体的复杂环境并提供可靠结果,但面对上亿种配体时,全面实验评估所有药物并不现实。相比之下,虚拟筛选将分子对接、虚拟化合物库与生物靶标的结构数据相结合,通过高通量计算评估化合物与目标靶点相互作用强弱的成本则更具可行性。虚拟筛选主要有2种方法,基于配体的虚拟筛选(ligand-based virtual screening,LBVS)和基于结构的虚拟筛选(structure-based virtual screening,SBVS)。
当靶点信息匮乏但有已知有效药物时,一般可采取基于配体的虚拟筛选策略,如定量构效关系分析或药效团建模等方法。而当疾病靶点蛋白明确且其三维结构及结合位点信息已知时,基于结构的筛选策略通常是首选。在蛋白质三维结构预测技术发展之前,这类结构数据主要来自NMR或X射线晶体学实验。而如今,诸如AlphaFold、MEGA-Fold和RoseTTAFold等AI预测方法也能为SBVS提供蛋白质三维预测模型,有效填补了实验数据缺失的空白。随着蛋白质三维结构预测技术的进步,越来越多的蛋白质结构信息被获取,有力推动了SBVS的发展。例如,Weng等曾利用AlphaFold预测了当时结构未知的WSB1蛋白三维构象,并基于该模型筛选获得了具有高潜力的配体化合物。
在SBVS中准确预测和理解蛋白配体相互作用能够加速药物研发进程,优化药物分子结构,并揭示生物分子的功能机制。近年来随着深度学习模型的应用,蛋白配体相互作用的研究取得了显著进展。蛋白配体相互作用预测主要有三类方法。
一类主要偏重互作结构的预测,代表性方法包括EquiBind、TANKBind、DiffDock、RoseTTAFold All-Atom和AF3等。这类方法的优点是可以获得蛋白质与小分子复合物的较为精确的结构,但是这类方法普遍速度慢,不适用于高通量虚拟筛选任务,并且也不会给出结合能的评估。
第二类方法侧重于结合能的预测,如GraphDTA、PSICHIC、ΔVinaRF20、RTMScore和InteractionGraphNet等。这类方法推理速度快,但也存在泛化能力差或者需要大量构象采样、使用繁琐的缺点。
第三类方法就是在药物筛选中广泛应用的分子对接方法。除了传统的AutoDock、AutoDock Vina,近年来还诞生了GNINA、DSDP/DSDPFlex、RosettaVS、Interformer和SurfDock等融合了AI的方法。这类方法可以同时获得复合物结构和结合能,在SBVS中最为常用。
但是目前各类SOTA方法仍然需要针对性的改进以满足更广泛的实际应用需求。首先,大部分方法单独考虑结构预测任务与结合能预测任务,使得两个任务分离,而实际应用中往往需要能够准确预测结合能,同时输出可解释相互作用细节的结构信息,这就要求进一步开发结构预测和结合能预测一体化的综合筛选方法。其次,现有方法的性能评估数据集单一,测评功能单一,使得在实际应用中泛化能力差。最后,现有方法往往无法在精度和速度上取得平衡,因此在实际的SBVS应用中无法发挥作用。这就需要我们发展出快速采样联合多精度打分的策略,极限优化速度与精度。另外,药物分子的新颖性、选择性和可合成性是新药研发的关键,因此近年来研究者发展了各类分子生成与设计技术,通过结合AI、计算化学和合成生物学等方法,显著提升了药物研发效率。
2.3.3 药物分子从头设计
药物从头设计(de novo drug design)是一种基于靶点结构直接构建形状与性质互补的全新配体分子的技术。这一方法能够提出结构新颖且具有启发性的先导化合物,在药物研发过程中具有重要的原创性意义。现在药物分子的概念已不限于小分子药物,所以药物分子从头设计也分为小分子药物设计和(生物)大分子药物设计两大类。
(1)小分子药物设计。据估计药理活性化学空间中,我们可以找到的药物分子的个数是1060,如何在这样巨大的化学空间中进行高效搜索发现候选的药物分子呢? 分子生成式模型是一个极具前景的方向。生成式AI是AI的重要分支,其思想是试图学习训练数据的概率分布,提取有代表性的特征,产生一个低维的连续表示,最终通过从学习到的数据分布中采样来生成新的数据。近年来,由于Transformer和扩散模型(diffusion models)的发展,使得生成式AI在复杂分布上的表现得到极大提升,展现了强大通用性,已经应用在了文本生成(如GPT-4o)、图像生成(如MidJourney)和视频生成(Sora)等领域。2022年底OpenAI发布了ChatGPT,由于它能进行自然流畅的对话,因此引起了生成式AI的热潮。而生成模型的发展也为解决分子设计难题带来了新的思路,当生成模型应用于生成分子时,其本质是学习训练集中分子的分布,从而获得与训练集中的分子相似但不同的分子集合; 也可通过结合进化算法或强化学习等算法,生成具有特定生物活性或理化性质的分子。
分子生成模型可以根据其设计目标和实现方式分为两大类: 目标导向型和结构导向型。目标导向型模型通过优化目标函数(如药物活性、药代动力学性质等)来生成分子,通常采用强化学习或潜在空间导航技术,能够在无结构约束下优化分子。例如,REINVENT使用策略梯度方法对SMILES字符串生成模型进行微调,以生成符合特定目标的分子。DeepFMPO结合策略梯度和Q学习(Q-learning),利用“执行者-评论者”(actor-critic)方法实现最优分子设计。结构导向型模型则通过条件深度生成模型生成具有特定结构的分子,通常用于改进现有化合物的结构,以提升其性能。例如,Delete模型基于蛋白质结构和候选片段生成分子。分子生成模型通常基于以下几种深度学习架构: 变分自编码器 (variational autoencoder,VAE)、GAN和Transformer模型。条件变分自编码器(conditioned variational autoencoder,CVAE)和连接树变分自编码器(junction tree VAE,JT-VAE)模型属于变分自编码器,通过编码和解码过程将分子结构映射到隐空间,然后从隐空间生成新的分子。研究表明,分子的SMILES表示和图表示都可以被VAE编码和解码到隐空间中,在该空间中分子不再是离散的,而是可以解码回离散分子表示的实值连续向量; 不同向量之间的欧几里得距离将对应于化学相似性。GAN通过生成器和鉴别器的对抗训练生成分子,生成器负责生成新分子,鉴别器则判断生成的分子是否真实,如MolGAN模型。GenMol则是一个基于Transformer的通用分子生成模型,利用Transformer的强大编码能力支持从头生成和片段扩展。
虽然生成分子本身不是一项很复杂的任务,但是如何生成化学上有效、并表现出我们想要的特性的结构是一个具挑战性的问题。实现这一目标的最初方法涉及在现有数据集上预训练模型,然后将其用于迁移学习。通过校准数据集对模型进行调整以允许生成偏向特定属性的结构,之后可以使用不同的算法(如强化学习)进一步校准。然而这种方式在化学有效性方面存在困难,此外,依赖预训练数据集也会限制搜索空间并引入偏差。摆脱预训练的一种尝试是使用马尔可夫决策过程(Markov decision process,MDP)来确保化学结构的有效性,并通过深度Q学习来优化MDP以获得所需的属性。
扩散模型是近年来新兴的生成模型,在分子生成和分子设计领域取得了显著进展。这种模型在生成具有复杂几何结构和物理化学属性的分子方面表现优异,尤其在3D分子生成中展现了巨大潜力。扩散模型也具有灵活性和稳定性,可以通过条件生成特定属性的分子,而且与GAN相比,在训练过程中不依赖对抗性训练,避免了模式坍缩问题。尽管深度分子生成模型仍面临一些挑战,如合成可行性、数据质量与偏见以及多目标优化等,但使用AI探索化学空间已经显示出巨大的前景。它为我们提供了探索化学空间的新范式,以及一种新的检验理论和假设的方法。
(2)大分子药物设计。随着分子生物学与结构生物学研究的深入,科研人员在代谢通路解析、病理机制阐明以及大分子结构与功能研究等方面取得重大突破,使得大分子药物逐渐成为治疗复杂疾病的关键武器。相较于小分子药物存在的半衰期短、毒性较大、靶向性差及专利易被仿制等局限,大分子药物展现出显著优势: 特异性强、疗效显著、安全性高、半衰期长且仿制门槛高,尤其在复杂系统性疾病治疗领域具有不可替代性。在此背景下,大分子药物研发正迎来快速发展期,其发展势头已开始超越相对成熟的小分子药物研发体系。
核酸类药物(包括siRNA、mRNA、ASO、CRISPR系统等)通过直接调控基因表达实现疾病治疗,在肿瘤、遗传病和传染病领域展现出巨大潜力。然而,其开发面临序列设计复杂、递送效率低、脱靶效应显著等挑战。近年来,AI通过高通量数据建模与生成式设计,正在重塑核酸药物的研发范式。2023年百度团队开发了LinearDesign算法,使用动态规划将mRNA序列搜索空间从指数级降低到多项式级,仅需11min即可完成新冠mRNA疫苗序列优化。2024年他们又在LinearDesign算法基础上提出了基于神经网络的LinearDesign2设计算法,预测翻译效率都得到了明显提升。
另一类大分子药物则是蛋白与多肽类分子。设计具有定制结构和功能的蛋白质是生物工程的长期目标。最近,深度学习的进步使得蛋白质结构预测接近实验精度,这也促进了蛋白质设计的进步。蛋白质设计与蛋白质结构预测二者其实是一体两面的双生问题。众所周知,肽链会折叠成复杂的三维结构,这种三维结构以某种方式编码在构成肽链的氨基酸序列中。也就是说,氨基酸的线性序列决定了蛋白质的三维结构。因为这个重要发现,Christian Anfinsen在1972年被授予诺贝尔化学奖。这意味着原则上我们可以根据氨基酸序列直接预测三维结构。反之亦然,给定一个具体的蛋白质三维结构,理论上我们可以反推出构成这个蛋白质的氨基酸序列。这一正一反两个问题就是蛋白质研究的核心。
蛋白质是生命通过数十亿年逐渐进化而来的,它们就像微型机器人,在生命体中承担着各种各样的重要职能。但随着近年来人均寿命不断提高,人类面临着癌症和神经退行性疾病等全新的挑战。如果还是依靠大自然进化出全新的蛋白质来解决这些问题,恐怕要等上数亿年的时间。但如果我们能够按需设计出蛋白质,便能在短时间内取得突破性成果,这就是蛋白质设计的价值。但是一个典型的蛋白质包含100多个氨基酸构成的序列,而氨基酸本身就有20种,这就意味着潜在蛋白质序列组合有20100个,显然通过暴力计算无法完成这个任务。而蛋白质结构预测则为蛋白设计提供了强大基础,Baker等就以trRosetta结构预测网络为基础提出了一种蛋白幻想设计算法。该方法首先生成一段随机的氨基酸序列,并将其输入trRosetta结构预测网络,以预测起始残基-残基间距离。这一步显然不会生成任何有序结构。然后,他们在氨基酸序列空间对现有序列进行蒙特卡洛采样,并对网络预测的残基间距离分布与所有蛋白质的平均背景距离分布之间的对比度(KL散度)进行优化。以不同的随机序列作为起点进行优化,可以得到不同的、跨越多种序列和结构排列的新型蛋白分子结构,这一过程被称为网络幻想(network hallucination)。这项研究实际上从原理上证明了:为结构预测而训练出的深度神经网络,也可以被利用来进行蛋白质结构的从头生成和设计。
蛋白修复设计则是另外一类方法,如RFjointInpainting算法,其输入端是缺失的不完整蛋白骨架,含有部分序列和结构。输出端则是完整的骨架,缺失部分的结构和序列都被修复出来。近年提出的RF Diffusion则是一种蛋白质结构扩散设计方法,这种算法其实是受到了图像生成算法的启发,通过逐步去除噪声生成一个全新的蛋白质结构。RF Diffusion模型被证明非常适合各种蛋白质设计任务,只需在推理中添加对称化步骤并利用RF架构的SE(3)等变性,RF Diffusion就能够生成具有循环对称性和点群对称性的大型同源寡聚体组装体。此外,RF Diffusion能够构建对称的motif和非常小的motif,如来自酶活性位点的单个残基,这对于蛋白幻想或RFjoint几乎是不可能的。最后,该模型设计的蛋白质Binder仅以目标结构为条件,某些目标的湿实验室成功率高达50%。
蛋白质结构扩散设计的一个有趣的替代方案是扩散蛋白质序列。目前,离散变量(如氨基酸)的扩散性能比自然语言建模的自回归或掩码模型更差。然而,对于蛋白质来说,序列扩散可以比结构扩散简单得多,并且存在大量的蛋白质序列功能数据(结合或酶活性),这些数据可以潜在地用于训练模型以对序列执行分类器指导。这就是ProteinGenerator (PG)的初衷。PG基于RoseTTAFold结构预测网络,采用序列空间扩散模型,从噪声序列逐步去噪生成序列-结构对。该模型通过迭代优化序列和结构的联合分布,支持多模态约束(如氨基酸组成、二级结构)引导生成过程,显著提升了设计的灵活性和成功率。
另外一个重要的蛋白设计工具是ProteinMPNN144,它是一种基于深度学习的图神经网络,专门用于根据给定的蛋白质骨架结构预测其氨基酸序列。该模型通过利用蛋白质的进化、功能和结构信息,生成可能折叠成目标三维结构的氨基酸序列。ProteinMPNN基于消息传递神经网络 (Message Passing Neural Networks,MPNN)架构,其输入为蛋白质的三维结构(PDB格式),模型将其表示为图结构。模型通过节点(氨基酸)和边(化学键)的交互传递信息,更新节点和边的表示。通过随机解码顺序生成氨基酸序列,并利用位置耦合处理多链蛋白质。ProteinMPNN能够在几秒钟内完成序列设计,适合大规模蛋白质设计任务,在多个下游任务中的成功案例证明了该方法的巨大潜力。但是另一方面,对结构等信息的高度依赖等特征也对该方法的进一步发展提出了要求。
蛋白质设计领域正经历方法论整合的重要发展阶段,各类模型(包括序列模型、结构模型以及序列-标签模型等)的传统区分正在弱化。当前研究突破主要体现在三个维度: 一是实现了结构感知模型与高性能序列模型的有机融合; 二是创新性地引入了自然语言处理和计算机视觉领域的技术手段,从而获取更为全面的蛋白质表征;三是将生物物理原理融入机器学习框架,显著提升了模型的泛化能力。在技术应用层面,优化后的采样算法有效提升了生成序列的可靠性,而新兴的”自主实验平台”通过整合不确定性评估模型与实验流程,为缩短设计周期提供了新范式。展望未来,跨模态的统一设计框架将成为主流,这种整合方案将支持高效序列生成、复杂多目标优化,并最终实现具有超自然功能的全新蛋白质创制。
另外值得指出,AI不仅已经在药物发现的靶点发现、虚拟筛选和分子设计环节得到了广泛应用,近年来AI在药物递送领域的应用也取得显著进展。药物递送系统在优化药物的药代动力学以及药效学表现方面有重要作用,而AI技术可以赋能药物-辅料相互作用预测、配方优化、关键工艺参数预测及递送材料高效筛选等药物递送系统的关键环节,系统性推动药物递送研究范式发生变革,形成“数据驱动-模型预测-实验验证-临床转化”的新范式,突破传统药物递送系统研发周期长、成本高的限制。例如,王建新团队等通过“人工智能深度学习预测+实验验证”的交叉研究方法,高通量筛选天然产物分子库,发现了兼具脂质膜调控与葡萄糖转运蛋白1 (Glut1)靶向功能的天然化合物,并构建了新型双功能脂质体载药系统,在小鼠模型中展现出肿瘤靶向与治疗增效作用。中国科学院上海高等研究院的团队提出了一种多模态可解释性质预测模型,实现了快速精准预测mRNA脂质纳米颗粒(mRNA-LNPs)的转染效率,能够快速筛选出稳定有效的LNPs,提高mRNA药物递送效率,为多种疾病的个性化治疗提供了可靠的研究方法和工具。AI技术正通过精准预测、快速筛选和优化设计,加速药物递送系统的开发。而AI的重要性不仅在于优化了药物递送系统的技术细节,而是在于其重构了研究范式本身——从“试错科学”转向“预测科学”,从“静态优化”升级为“动态演化”,最终实现个性化的递送设计,为精准医疗和个体化治疗提供了强有力的技术支撑。
2.4 AI赋能精准医学
精准医学以个体化诊疗为核心, 通过整合基因组、表型组、环境等多维度数据, 实现疾病预防、诊断和治疗的精准化。传统的医疗模式往往依赖医生的经验和主观判断, 而AI通过强大的数据分析和学习能力, 以更精确的方式处理庞大的医学数据, 识别出人眼难以察觉的细节, 从而大大提高了诊断和治疗的准确性。近年来, AI技术成为推动精准医学发展的核心驱动力, 其应用已渗透至医学影像处理、生物标志物发现、药物研发、临床决策支持及健康管理等全链条。
AI在医疗领域最具突破性的应用之一, 是医学影像与数据的高效、精准分析。AI技术依托深度学习与机器学习算法, 能够高效处理海量复杂医疗数据, 精准识别传统诊断方法难以捕捉的细微特征, 从而避免由于高度依赖医生的临床经验与主观判断而造成的漏诊与误诊风险, 显著提升疾病诊断的时效性与准确性。2024年哈佛医学院联合斯坦福大学、布莱根妇女医院等国际顶尖科研机构, 在Nature杂志上发表了具有里程碑意义的癌症诊断研究成果——CHIEF模型。该模型采用先进的弱监督学习框架, 从海量病理图像中提取关键特征, 实现了对19种癌症的高效诊断, 准确率高达94%, 显著超越传统深度学习模型。与现有AI方法相比, CHIEF在整体性能上实现了36.1%的提升, 尤其在癌症检测、肿瘤基因变异分析及患者生存率预测等关键指标上表现卓越。通过智能分析病理图像中的关键区域, CHIEF不仅能够精准识别不同癌症类型, 还可预测与癌细胞生长密切相关的基因突变, 为精准医疗的临床实践提供了强有力的技术支撑。
除了提高诊断的精度, AI也能通过整合医学影像、电子病历、基因测序等数据, 对患者个体数据进行深入分析, 从而在更全面地理解患者病情的基础上辅助医生设计更加个性化的诊疗方案。特别是, 随着大语言模型的兴起和能力的不断提升, LLM在医学领域的推理能力已实现质的飞跃, 部分任务表现甚至超越人类专家。例如, OpenAI的o1-preview模型在The New England Journal of Medicine143个临床病理学会议(CPCs)病例测试中展现出卓越性能: 整体诊断准确率达78.3%, 远超传统大语言模型和临床医生水平。特别在鉴别诊断、诊断推理和管理推理三个关键维度, o1-preview表现出接近专家级的判断能力。更引人注目的是, 该模型在后续检查方案推荐方面达到87.5%的准确率, 证实了AI在复杂临床决策中的实用价值。最近, 浙江大学也开发了AI病理助手OmniPT, 整合了视觉识别与自然语言处理技术, 实现了病理图像的智能化快速分析, 能够在1~3s内精确定位癌症病灶区域, 诊断准确率突破95%。在临床应用方面, OmniPT在胃癌、结直肠癌及宫颈癌等多种恶性肿瘤的诊断中展现出卓越性能。通过其独特的多任务协同分析机制, 可同步完成癌症分类、病灶分割及病变检测等多项关键任务, 显著提升了病理诊断的效率与准确性。
由于药物疗效和毒性的个体差异显著(如治疗窗窄、不良反应多), 传统群体药代动力学模型在精准用药中存在局限性。AI能处理高维、非线性数据, 挖掘真实世界用药数据中的潜在规律, 更准确地预测血药浓度和剂量, 优化用药方案。例如, Huang等收集了407例接受静脉注射万古霉素的儿童患者的血药浓度监测数据, 以万古霉素谷浓度为预测目标变量, 筛选出了5种具有更高相关系数的机器学习算法构建集成模型, 并获得了最优预测效果。研究表明, 与传统药代动力学模型相比, 该机器学习模型具有更好的拟合效果和更高的预测准确度。该集成模型可用于万古霉素血药浓度预测, 尤其适用于个体差异显著的儿童患者群体。
基于表型组数据的真实世界应用也是精准医学的重要一环。基于表型的药物发现(phenotypic drug discovery, PDD)是对基于靶点的药物发现的重要补充, PDD采用与靶标无关的方法, 专注于化合物在疾病相关生物系统中的表型效应。这一策略利用已标注作用机制的参考化合物, 来揭示测试化合物的作用机制。迄今为止, PDD在首创新药的发现方面已做出重要贡献。PDD也是天然产物发现的主要方法, 是识别新靶点和作用机制的基础。AI可以高效地综合分析多维度的人体药物反应数据, 如药代动力学(pharmacokinetics, PK)数据, 药物在不同个体中的吸收分布、代谢和排泄(absorption, distribution, metabolism, and excretion, ADME)参数, 药效动力学(pharmacodynamics, PD)数据, 不同剂量药物对靶点、细胞和整体生理系统的影响, 电子健康记录(EHRs), 临床试验数据等, 因此, AI在表型驱动的药物发现中具有重大的应用价值, 能为精准医疗和生物医学研究的发展开辟全新路径。例如, 郑明月课题组开发了基于自我监督表示学习的深度生成模型TranSiGen, 能够通过分析细胞基因表达和化合物分子结构, 高精度重建化学诱导的转录谱, 从而捕获细胞和化合物之间的复杂信息关联。该模型在配体虚拟筛选、药物反应预测和药物再利用等下游任务中表现优异, 尤其在胰腺癌药物发现中的应用得到体外验证, 展示了识别有效化合物的潜力。基于表型数据和AI模型, 也可以建立健康预测与早期预警系统, 通过多组学信息分析, 开发能够评估个体健康风险的预测模型, 涵盖疾病发生的早期预警。例如, 基于基因信息、生活方式、环境暴露等因素, 预测心血管疾病、糖尿病和癌症等的发生风险。根据个体的表型数据, 也可以利用AI开发定制化的健康管理方案, 特别是在慢性病管理、老龄化社会中的老年人健康管理等方面。通过个性化干预, 帮助提高人群健康水平, 减轻社会医疗负担。更大的层面上, 基于表型组数据可以构建大数据AI分析平台, 帮助政府进行科学的公共卫生决策。例如, 通过疫情数据的实时监控和预测模型, 优化防控策略, 提高应急响应能力。
2.5 AI赋能绿色生物制造
绿色生物制造是以生物合成化学、合成生物学、基因编辑、人工智能等前沿技术为核心,利用酶催化反应或通过改造微生物/生物系统实现低碳、高效的生物基材料、化学品、药物等的高效合成与生产。其核心目标是替代传统高污染、高能耗的化工工艺,推动工业、农业、医药等领域的绿色转型。在全球生物经济迈向高质量发展的关键时期,国务院将生物制造列为未来产业,强调以科技创新为引擎推动产业升级,全力构建绿色可持续的生物制造体系。各地方政府也纷纷出台合成生物产业专项政策,建设“AI+生物制造”创新联合体。
“生物合成化学”与“合成生物学”是生物技术与化学交叉领域中的两个重要研究方向。生物合成化学重点关注生命活动中物质的生物合成机制,进而利用生物体系、生物元件等完成特定化学反应、合成特定目标分子或新功能分子。生物合成是20世纪末随着生命科学的发展而出现的合成方法,对比化学合成,生物合成可以实现高选择性、高反应性和高经济性,尤其在手性中心的构筑、惰性碳的活化以及复杂天然产物的合成方面具有极大优势。但是,现阶段生物合成也存在酶元件少、酶开发难和细胞工厂设计构建难的瓶颈。这些困难使得生物合成的灵活性远低于化学合成,生物合成反应还很难像化学合成一样进行任意设计,提高灵活性是当前生物合成研究的重要目标。因此,生物合成化学利用在化学合成领域成熟的化学反应机制研究方法,探索生命体系中物质的生物合成机制就显得格外重要。在生物合成化学中充分结合AI和理论模拟计算来进行生命体中化学反应机理研究,厘清生物合成机制的基础上可以指导对于生物酶的改造以实现特定的化学反应的催化。例如,2018年诺贝尔化学奖获得者Arnold教授,在理解了生物酶催化机制的基础上,通过改造P450酶中与血红素共价的第五配基可以使该酶催化C–B和C–Si键的合成反应。AI技术的引入进一步加速了这个方向的研究进展。
合成生物学的核心技术原理是对生物系统进行工程化设计和改造,通过对生物元件(如基因、蛋白质等)进行设计、组合和优化,构建出具有特定功能的生物系统。合成生物学采用的工程设计原理和工程学的可预测性来控制复杂生物系统,形成了一个以“设计-构建-测试-学习”(DBTL循环)为核心的研发模式。然而,合成生物学面临着一个巨大的挑战: 我们对生物系统的预测能力远远不如对物理或化学系统的预测,这就造成了我们对产生相关生物表型的底层机制理解不足,从而使我们在实践层面无法精确地按照特定要求设计生物系统。
AI技术的出现,为合成生物学提供了所需的预测能力,可以应用于合成生物学过程的各个环节。首先,在催化元件的发现、设计和改造环节AI展现了强大的潜力,其可以被应用于蛋白质功能和酶活性注释、酶的从头设计、酶的选择性改造以及酶的稳定性改造等。虽然UniProt数据库中已收录了约2.5亿条蛋白质序列,但其中约99.7%的蛋白质序列缺乏功能注释。如果能够准确地为这些蛋白质添加功能注释,就可以获得大量多样化的候选对象作为酶工程的起点加以探索。利用机器学习分类模型可以全面整合蛋白质序列与结构特征,从而能够更精确地预测蛋白的具体功能。最近,美国伊利诺伊大学香槟分校的研究团队开发了一种名为基于对比学习的酶功能注释(contrastive learning enabled enzyme annotation,CLEAN)的机器学习算法,通过对比学习框架能够对未经研究的酶类实现准确、可靠且高灵敏度的酶功能预测,准确率达87%,远超传统方法(40%)。酶功能注释工作未来的一个重点是对于混杂活性的标注,混杂活性既可能表现为对新底物保持相似的化学反应,也可能表现为完全不同的反应类型,通常是酶进化出非天然活性的起点。而混杂活性难以检测,或尚未经过实验验证,因此通过实验测定来更新酶功能数据库至关重要。
此外,借助基于生成式预训练架构的大型语言模型(LLM)对文献进行文本挖掘,也有望通过提取尚未纳入现有数据库的科学文献知识,发现缺失标签并更新已有数据库。尽管通过注释已知蛋白质序列可以发现许多功能性酶,但生成自然界中从未出现过的全新序列同样具有重要意义——这些序列可能带来前所未有的性质组合,甚至催生出非天然活性。AI凭借其强大的数据驱动学习能力和在未知空间的探索能力正在帮助科学家们以前所未有的精度和效率设计蛋白酶,并达到提高产量和改进功能等目标。总的来说,蛋白酶的AI设计方法可分为两大类: (1) 纯序列生成; (2) 结构设计,即寻找能够折叠成特定结构或骨架的序列。
在纯序列生成方面,蛋白质语言模型(PLMs)可通过给定已知酶家族作为条件,生成具有该功能的新序列,而无需直接考虑结构。例如,Naik等开发的ProGen模型能够在大规模蛋白质家族中生成具有可预测功能的蛋白质序列,针对五个不同溶菌酶家族进行微调生成的人工蛋白的催化效率与天然溶菌酶相当,而与天然蛋白的序列同源性最低仅为31.4%。Zelezniak等基于生成对抗网络模型提出了ProteinGAN模型,直接从生物序列中学习潜在的氨基酸关系,并产生具有天然生化特性的新的功能蛋白序列,在对苹果酸脱氢酶(MDH)进行序列设计时,显示出了24% (13/55)的设计成功率。而在结构设计方面,则可以利用2.3.3小节中介绍的蛋白质结构生成和设计工具直接设计所需的酶骨架。例如,Kao等就采用ProteinMPNN设计了序列发散的泛素变体,这些变体对E3泛素-蛋白连接酶Rsp5外部位点的HECT结构域具有高亲和力,并且在这些变体中鉴定出了几个具有更高的蛋白质产量、保持高热稳定性和增强的结合亲和力的成功设计。最近,Baker团队利用AI从头设计了具有复杂活性位点的丝氨酸水解酶,其能够加快一个四步化学反应,并且催化效率是之前设计的水解酶催化效率的6万倍,在酶工程领域具有里程碑意义。
除了可进行蛋白酶设计,AI也可以促进蛋白酶的改造和定向进化。例如,司同团队整合机器学习与贝叶斯优化算法指导自动化实验迭代,实现了蛋白质突变空间的高效探索,成功将鼠李糖酯合酶RhlA进行了改造,使其对C8底物的特异性提高了4.8倍。Alper等则采用基于3D卷积神经网络架构的MutCompute算法,成功对PETase塑料降解酶进行了工程化改造,开发出高效变体Fast-PETase。实验数据显示,该酶在50℃条件下仅需48h即可将未处理的聚对苯二甲酸乙二醇酯(PET)包装材料降解,更突破性地实现了塑料降解产物的再聚合,为塑料污染治理提供了创新解决方案。洪亮团队开发的Prime模型可以通过小样本干湿迭代在≤100个湿实验数据,数月内实现多款蛋白质的定向进化,部分蛋白产品已经落地产业化。除了酶工程,AI还可以优化整个代谢途径,甚至是复杂的生物回路系统,在代谢工程领域展现出了强大的应用前景。
AI驱动的代谢通路发掘与优化是近年来合成生物学和代谢工程领域最活跃的交叉前沿之一。由于细胞代谢网络具有高度复杂性,传统设计方法通常依赖文献检索、代谢建模和启发式分析,这些方法受限于计算吞吐量,难以从海量的代谢反应及调控网络中高效筛选最优改造靶点。AI驱动的集成建模方法为这一挑战提供了新思路,其能够在代谢网络建模中同时整合动力学特性、调控机制、替代模型结构及参数集合等多维因素。机器学习平台作为高通量分析工具,也日益广泛地应用于大规模代谢数据筛选,推动数据驱动的生物合成途径优化与微生物产能提升。例如,EcoSynther平台利用支持向量回归和前馈神经网络,仅两轮DBTL即将柠檬烯产量提升60%以上; BioAutoMata平台用贝叶斯优化番茄红素途径,产量比随机筛选高77%;这些研究都验证了AI在代谢工程中的高效优化能力。合成生物学领域内,人们在掌握目标产物的代谢路径后,需要进行基因层面的设计,利用CRISPR等基因编辑技术将目标基因导入,以实现目标产物的表达。基因回路的设计对于精确的基因调控至关重要。目前,AI工具在这一过程中扮演着越来越重要的角色。下一步,科学家需要开发DNA-蛋白多模态预训练模型,并基于此研发蛋白质改造、代谢通路改造、底盘菌改造的干湿迭代的小样本学习方法,彻底颠覆合成生物学靠专家经验+大量湿实验试错效率低下的研究范式。取而代之的是AI主导、配合少量湿实验就能实现合成生物学全场景的应用,包括单个生物元件(如酶)、代谢通路,乃至底盘细胞的快速设计和优化,大大加速生物相关产业源头产品研发。
03 AI赋能的化学生物学中的挑战与展望
AI赋能的化学生物学作为新兴研究范式,在加速科学发现与技术创新方面展现出巨大潜力,但也面临多维度的挑战,包括数据瓶颈、算力消耗、算法效率、模型泛化性、模型可解释性等多个层面。
AI模型的训练需要大量高质量的数据,而在化学生物学这类交叉学科研究中,数据往往具有稀缺性和不均衡性,这必然会限制AI模型的表达能力和泛化性。而且化学生物学不同实验技术、不同研究领域的数据在模态、格式和标准上千差万别,这导致了数据整合困难。例如,医学影像数据与基因组学数据就难以直接对接,需额外开发多模态融合模型,增加了算法复杂度。因此,在AI赋能的化学生物学领域,首先要建设生物分子、药物分子的可信数据空间,产、学、研、用联动,打通数据保护、开发、流通、交易的生态。在此基础上,建立和完善多组学标准化、规范化的大规模数据库。确立数据库的基本组织形式和提供的基本数据类型,并建立自动化的数据处理流水线。确立标准化、规范化的实验测定流程与标准,保证不同实验之间测定数据的可比性。进一步,将大语言模型接入数据库中完成基于自然语言描述的数据自动分类与处理,实现端到端的处理原始实验数据,将原有数据结构化存储,便于进一步设计多模态大模型,并能帮助数据提交者分析总结数据,具有寻找相关数据集的能力。另一方面,虽然化学生物学研究中已发展了多种高通量实验手段,但是按照传统模式内生的数据,仍然无法满足机器学习的需求。针对数据不足的痛点,人们已经开始尝试将AI和自动化、机器人技术相结合,从而实现从设计、实施到测试整个流程都不需要人干预的智能化自动实验系统。在这个领域,英国格拉斯哥大学、英国利物浦大学、美国麻省理工学院、美国伊利诺伊大学以及我国的北京大学、中国科学技术大学等均取得了重要进展。
智能化自动实验系统能够更有效、更便利地获取有价值的数据,可以成为智能化的生产科学数据的基础设施。当然在实验室自动化和智能化方面,还有很多方案和路径的细节需要讨论。例如,在自动化系统中是否有更简单且可靠的方式解决众多模块物理空间连接的问题; 将每一个新生成的实验结果放到之前的数据中进行训练,如何平衡模型的拟合能力和泛化能力; 机器寻找优化方向的强化学习算法如何进一步改进等。最近,LLM推理能力的不断增强为智能化自动实验系统的设计与开发提供了足够强的技术基础,以LLM模型为底座的多智能体(agent)架构可以将不同的专业模型和工具协同起来,由任务管理智能体分解任务并自主规划和调配各种工具来完成任务,从而实现完整科研闭环的自主运行,降低专业工具使用门槛,极大减少人为干预,提升实验效率和科研创新性。
近年来LLM的发展不断证明AI中存在尺度定律(scaling law),即模型的智能表现随着规模(如模型大小、数据量、计算量)的增加出现跃升,也就是“智能涌现”。因此,除了需要大量数据,AI模型的训练对计算资源的需求也越来越高。例如,GPT-3有1750亿个参数,训练用到了1024张英伟达A100芯片,而GPT-4模型的训练则达到了万卡的规模,且训练模型所用芯片也从英伟达A100更新到英伟达H100、B200,xAI最近发布的Grok3的训练甚至用到了20万张H100。这么巨大的算力资源消耗,对于科研机构来说是不可能负担的。特别是随着全球科技竞争加剧,美国、欧盟及英国相继宣布了对特定商品的贸易禁令,使得我国的科研工作者难以获取国外先进的计算芯片,这进一步加剧了算力需求的挑战。不过,我国人工智能企业DeepSeek近期发布的DeepSeek-R1模型成功实现了低算力条件下与国际前沿模型体系(如ChatGPT-o1等)的性能对标,这无疑证明了在没有大量最顶尖算力的条件下通过算法创新和工程优化,完全有可能达到甚至超过国际前沿模型的性能。所以在化学生物学领域的AI算法创新上我们仍有很多工作要做。另一方面,我们也需要同步建设国产科学计算和智能计算软硬件生态,促进算力基础设施的国产化替代。
数据驱动的AI技术虽然擅长通过对多模态复杂数据的整合进行预测和决策,但是在安全要求高的领域,如医疗诊断,我们必须要知道模型做出决策的原因,否则医生难以信任其建议。AI模型作为科学研究的工具,也需要可解释性来揭示其中的内在原理,但是深度学习模型的“黑箱”特性使得科学界对AI结论的接受度受限。可解释性对于质疑、理解和信任人工智能系统至关重要。可解释性还反映了我们的领域知识和社会价值观,为科学家和工程师提供了更好的设计、开发和调试模型的方法,并有助于确保人工智能系统按预期工作。可解释人工智能 (explainable AI,XAI)作为一个新兴研究方向,致力于使AI系统的决策过程更加透明和可理解。可解释人工智能正在从传统的特征归因方法向更高层次的语义解释发展。大语言模型和视觉-语言模型的引入为XAI带来了新的机遇,使得生成更自然、更有意义的解释成为可能。然而,解释的忠实性、稳定性和效率等基本问题仍需要进一步研究。未来的研究应该着重于: 可解释性和可靠性的提升、降低计算开销、增强与领域知识的结合、完善评估体系。随着技术的不断进步,可以预期XAI将在提高AI系统透明度和可信度方面发挥越来越重要的作用。
针对化学生物学领域的创新AI模型开发是需要关注的最重要方向。正如前文已做过的介绍,已有大量基于机器学习和深度学习方法解析生物谱学图像、分析多组学数据、理解基因调控的基础模型被开发出来。另一方面,高通量测序技术推进生命科学研究进入到“组学时代”,在海量组学数据支撑下,scBERT、scGPT、scFoundation、BioMedGPT和Evo2等大规模预训练模型被开发出来,并在医学和生物学多个领域取得了显著成功。这一系列的研究进展给了科学家们挑战物理与计算模型极限的信心。
2024年12月,斯坦福大学及多家国际顶尖机构的研究团队联合在Cell杂志上发表了题为“How to build the virtual cell with artificial intelligence: Priorities and opportunities”的综述论文,提出了人工智能虚拟细胞(AI virtual cell,AIVC)的概念,并且描绘了构建这一模型的思路蓝图。细胞是生命的基本单元,深刻理解其运作机制对揭示生命本质具有重要意义。智能虚拟细胞技术的出现有望引领生物学研究范式的革命性变革,推动高精度生命系统模拟进入全新发展阶段(图4)。这项突破性技术将重构科研范式——研究者不再局限于传统“假设-验证”模式,而是通过智能系统主动生成可验证的科学假说。尤其在处理海量生物数据时,虚拟细胞模型能高效缩小机制研究的假设空间,大幅提升发现关键调控因子的效率。虽然这类模型未必直接揭示因果关系,但其作为“假设过滤器”的价值已毋庸置疑。可以预见,智能虚拟细胞技术必将成为人工智能与生命科学交叉领域的战略高地,并将引发全球范围内的科研竞速。在化学生物学已具有较好研究基础的情况下,我国更需要积极应对历史机遇,加速在智能虚拟细胞方面的研究布局,建立根植于分子描述的多尺度、跨模态、可扩展、智能化的数字虚拟细胞(组织)模型,努力在AI赋能的化学生物学发展阶段获取主动(图4)。

图4 基于分子描述的多尺度、跨模态、可扩展、智能化的数字虚拟细胞模型的架构示意图
因篇幅限制, 加之相关领域进展十分迅速,本文中我们仅选择了几个有代表性的方向进行了讨论。在本文中,我们尝试对化学生物学研究中可能涉及的不同尺度和不同模态的深度学习相关的分子模型及其应用研究分别进行了简短的讨论。我们相信这些相对分散的研究虽然目前各自仍然只是在相对孤立的应用场景下进行,但是随着大语言模型和多模态任务的逐渐统一,具有跨尺度能力的预训练模型的发展将会为更加系统的、从分子到细胞甚至组织的定量研究提供基础模型和可交互的分子表征。基于预训练和生成式模型的分子表征不但可以统一不同模态(如分子设计和模拟、调控网络分析和药物效应研究)的计算任务,也可以为干湿闭环的智能化实验研究提供基础。我们相信整合多尺度和多模态信息的科学大模型具有产生涌现能力和带来化学生物学研究变革的潜力。当前,AI正在从单纯的研究工具逐渐转变为科学研究的核心组成部分,并正在形成全新的科研范式。另外,如果我们仔细思考AI能这么快地迭代发展的原因,或许这些成功经验能带给科学研究者更重要的启示,如AI领域开放社区的活力、规模化的生态等。因此,不久的将来”AI for science”这一重点强调AI工具性质的词汇或许将不足以代表AI与科学的交汇,而一个AI原生的科学生态圈(AI-cultured science EcoSystem,AICSES)可能将会是科学工作者和AI研究人员共同追求的目标。
参考资料:
Yang L, Gao YQ, Huang Y, Yang J. AI-empowered chemical biology. Sci Sin Chim, 2025, 55, doi: 10.1360/SSC-2025-0169
文章改编转载自微信公众号:智药邦
原文链接:https://mp.weixin.qq.com/s/SWxZrMux5H6wBheb6zj-Rg | 





















