随着量子力学在2025年迎来其百年纪念,机器学习原子间势(MLIP)已逐渐发展为分子建模领域的变革性工具,在量子力学精度与经典计算效率之间搭建起一座关键桥梁。2025年12月22日,卡内基梅隆大学的研究人员在《Nature Computational Science》上发表文章,题为“Machine learning interatomic potentials at the centennial crossroads of quantum mechanics”,由Olexandr Isayev担任通讯作者,对这一快速发展的领域进行了系统梳理与前瞻性展望。

文章从四个核心挑战出发,回顾并分析了MLIP的发展脉络,分别是如何实现化学精度、如何维持计算效率、如何提升模型的可解释性与可信度以及如何实现跨体系的普适泛化能力。这些进展共同勾勒出一条清晰的发展路径,指向具备预测能力、良好可迁移性且物理上自洽的机器学习框架,为下一代计算化学方法奠定了重要基础。
背景
2025年标志着现代量子力学(QM)奠基性发现问世一百周年,这一里程碑如今被正式纪念为国际量子科学与技术年。在回顾量子力学百年辉煌成就的同时,量子化学领域正经历着另一场深刻变革,即人工智能(AI)与机器学习(ML)向传统计算科学的深度融合。这些以数据驱动为核心的方法正在重塑分子模拟、化学性质预测以及基本理论框架中近似方法的构建方式。图1系统梳理了过去一个世纪中量子化学与人工智能领域的重要里程碑,展现了两条技术发展脉络逐步交汇的历史进程。
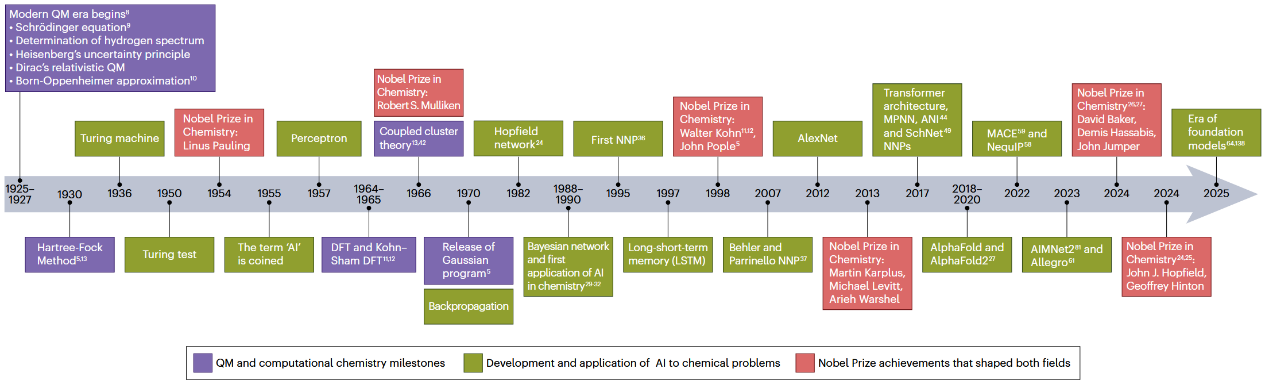
图1 量子力学百年演化时间线及同期人工智能事件,并标注人工智能在量子化学中的关键里程碑。
尽管早在1995年就已经出现了最初的神经网络势(NNPs),但现代意义上的MLIP通常被认为始于Behler与Parrinello于2007年提出的开创性工作。MLIP是一类利用机器学习近似原子体系势能面(PES)的计算工具,其根据原子的位置和化学身份预测原子间相互作用。Behler与Parrinello首次提出了基于原子中心对称函数的高维势能面通用神经网络表示方法,由此开启了一种全新范式,旨在弥合长期以来存在于量子力学精度与经典计算效率之间的鸿沟。
如今,MLIP已发展成为一个方法体系极为丰富的研究领域,不同模型从不同角度应对精确描述原子间相互作用这一核心挑战。随着应用场景不断拓展,研究人员也面临着更加复杂的权衡问题,需要在精度、效率、可解释性和泛化能力等多个目标之间寻找平衡。因此,本文围绕上述四个相互关联的关键主题,对MLIP的当前进展与未来发展方向进行了系统性的总结与展望。
追求化学精度
在能量预测中实现所谓的化学精度(通常定义为 1 kcal mol⁻¹)长期以来一直是量子化学与原子尺度模拟的核心目标。诸如杂化密度泛函理论(DFT)或双杂化DFT等方法,其计算复杂度分别随原子数按O(N³–N⁴)或O(N⁵)标度增长,在实际应用中往往能够提供足够合理的精度。而耦合簇理论,最常用的是在完全基组极限下的CCSD(T),其计算复杂度高达O(N⁷),可逼近化学精度这一黄金标准,但对于大体系而言,其计算代价往往难以承受。MLIP则有望在计算速度提升数个数量级的同时,提供接近从头算的精度。图2从整体视角展示了MLIP体系结构从手工构造描述符到现代等变方法的演化脉络。
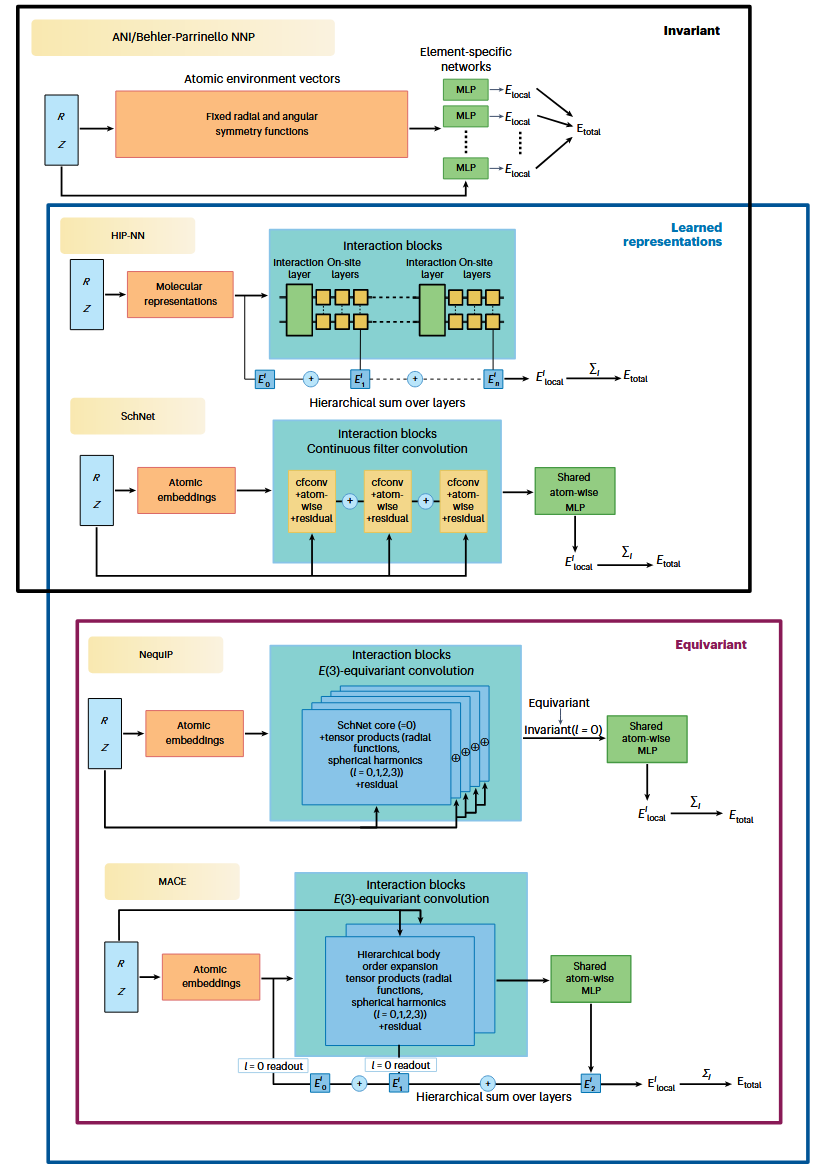
图2 MLIP体系结构的演化
MLIP所能达到的化学精度,是相对于用于生成训练数据的参考方法而言定义的。尽管在表示方式与体系结构层面上的改进已显著提升了模型性能,但当前MLIP的诸多问题与潜在陷阱,往往可归因于底层理论本身的误差,或高质量数据库的缺乏。为缓解高精度数据稀缺的问题,研究者广泛采用了多保真训练、Δ学习、迁移学习以及元学习等策略。这些方法通过学习误差修正而非绝对物理量,或从大规模低保真数据集中迁移知识,从而绕开高精度数据不足的瓶颈。
对计算效率与可及性的追求
从头算方法的计算代价通常随体系规模呈多项式标度增长,而对高精度方法而言,其计算需求会急剧上升。MLIP在计算化学方法谱系中占据着独特位置:相较于量子力学(QM)方法,它们能够显著降低计算成本,同时在速度上可与传统力场相媲美。然而,在MLIP领域中同样存在效率与精度之间的经典权衡问题。要实现对这一权衡的理解与优化,需要系统分析主导计算开销的关键因素,包括神经网络复杂度、随体系规模的标度行为,以及力和Hessian的计算代价,并在模型复杂性与实际计算性能之间作出审慎取舍。
等变体系结构在数据效率和跨多样化学环境的可迁移性方面展现出卓越性能。这类模型在需要对多种分子构型实现高精度预测的应用中尤为突出。不变型消息传递神经网络在保持强表示能力的同时,通常具有更为有利的线性标度特性,这是通过对局域原子环境的有效描述实现的。因此,即便等变网络日益受到关注,研究者仍持续改进不变型MLIP,通过引入基于物理的组成要素来提升泛化能力,同时维持计算效率。AIMNet系列MLIP即是此类领域知识驱动且计算高效模型的代表。为应对计算效率方面的挑战,研究者还探索了基于注意力机制的体系结构。注意力机制在捕获长程相互作用方面表现出强大能力,但其计算复杂度通常随体系规模呈二次增长。代表性方法包括So3krates以及EScAIP。
当前的等变型MLIP,尤其是NequIP和MACE等体系结构,还面临着严重的内存标度限制,使其应用范围被限制在相对较小的体系中。相比之下,不变型模型已被常规性地扩展至包含数十万原子的体系(图3c)。为克服这些可扩展性限制,研究者提出了多种解决方案。例如NVIDIA推出的cuEquivariance通过基于CUDA加速的构建模块来应对等变模型的标度瓶颈。知识蒸馏则提供了另一条可行路径,通过让复杂的等变“教师”网络训练轻量化的“学生”模型,在保持预测精度的同时,大幅降低计算成本和内存需求。此外,诸如量子力学/分子力学(QM/MM)等混合方法也可能成为应对纯ML模型可扩展性问题的有效替代方案。
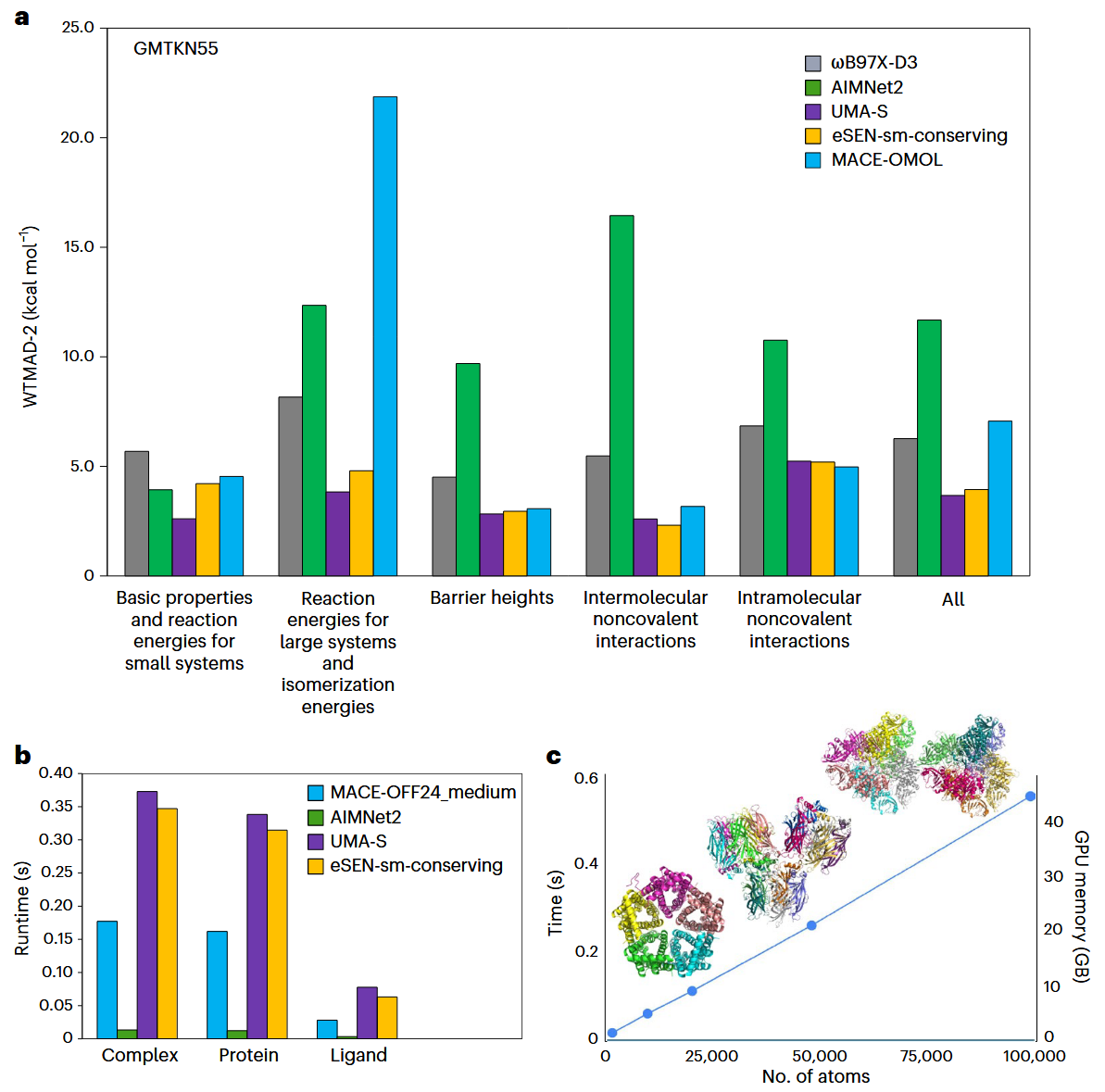
图3 MLIP的精度与尺度对比
可解释性与可信性的挑战
可解释性或许是现代MLIP中最为持久且棘手的问题。与具有明确物理含义参数的经典力场不同,MLIP学习得到的表示通常缺乏直接的物理解释,使得人们无法从中提取诸如键的性质、电荷转移效应或配位偏好等具有化学意义的洞见。现代MLIP体系结构往往包含数以百万计的可训练权重,其高维参数空间使得对单个参数进行有意义的分析或建立直观的物理理解几乎不可能。然而,MLIP的目标并不一定是复现人类对化学现象的理解方式,而是提供在物理上自洽且精确的预测,并严格遵守能量守恒、平移不变性以及合理化学行为等基本约束。
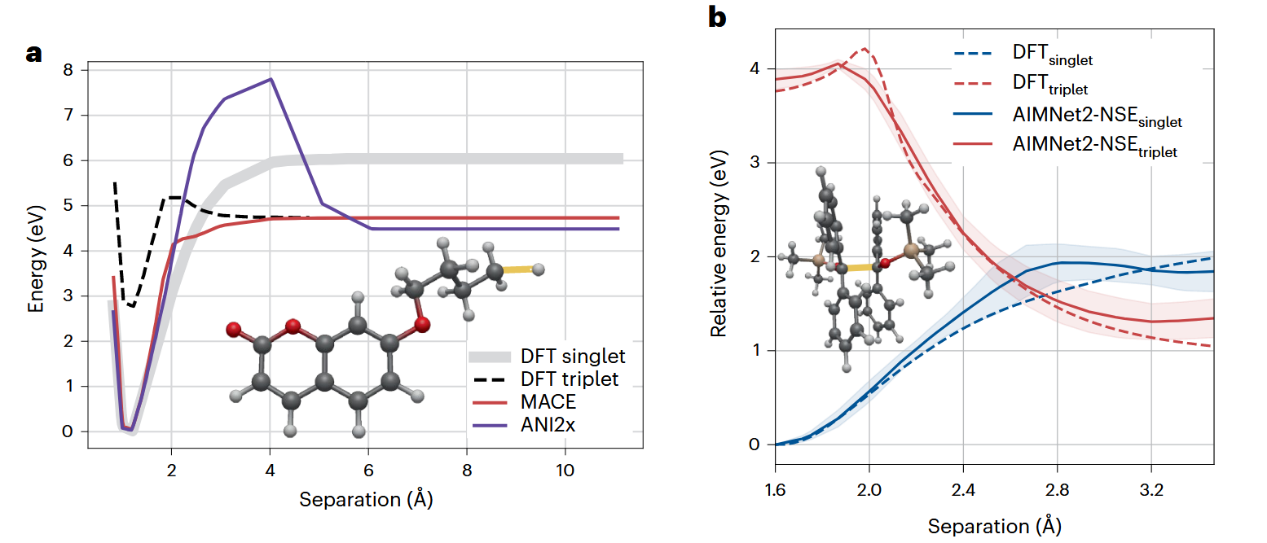
图4 MLIP在开壳层键解离问题中的性能
贯穿MLIP发展历程的一个关键认识在于通过领域知识驱动的表示,将物理学先验引入机器学习模型至关重要。诸如能量守恒、光滑性以及各种对称性等约束,可以通过体系结构设计显式地加以保证,也可以通过损失函数惩罚项与训练数据选择的方式隐式引入。针对特定电子结构复杂性的处理需求,可以设计专用的MLIP体系结构。随着新数据集的不断涌现以及数据整理与筛选质量的提升,最新一代MACE模型已将电荷与自旋作为额外输入特征嵌入网络,并将其视为传递给模型的外部标签。此外,近期研究表明MLIP不仅能够识别新的反应路径、预测出人意料的成键模式,还能为复杂催化机理提供深入见解。然而,要充分释放这一潜力,还需要配套的高级分析工具,以从计算结果中提取具有化学意义的洞见。将可解释的物理模型与机器学习组件相结合的混合方法,被认为是一条极具前景的发展方向,有望在兼具AI的精度与灵活性的同时,保留传统方法的可解释性与物理直觉。
迈向通用基础模型
第四个也是最具雄心的目标是通用泛化能力,这一目标正推动当前研究向基础模型方向发展。基础模型受到大语言模型的启发,旨在构建能够在多样化化学环境中、针对任意元素组合都保持高精度描述的通用原子间势。这类模型应当遵循规模定律,即随着模型规模、训练数据量和计算资源的增加,其性能能够持续提升。真正意义上的基础MLIP将消除对特定体系的参数化需求,使复杂、多组分体系的无缝探索成为可能。然而,在覆盖极其广阔的化学空间的同时,仍然保持高精度与高效率,带来了前所未有的挑战。
展望
在量子力学诞生一百周年之际,人工智能与量子化学的深度融合正站在一场革命性飞跃的门槛上,这一飞跃将从根本上重塑分子科学的研究范式。未来十年,有望打破长期制约计算化学的边界,开启一个精度与效率趋于统一、可解释性通过物理启发方法持续提升的新纪元。随着AI技术的不断进步,其与自然科学的融合将开启全新的研究前沿,从根本上改变人类理解与操控自然世界的方式。
在未来几年内,预计将出现真正意义上的通用型MLIP,其训练数据规模将超过十亿个分子构型,覆盖整个元素周期表。这类基础模型将成为化学领域的“GPT-4时刻”,标志着研究工具从高度专用化向通用化化学智能的转变。
新一代模型架构的出现,有望解决当前在高精度等变模型与高可扩展不变模型之间不得不做出的权衡。通过设计保持线性标度的稀疏等变网络,在保留几何感知能力的同时显著降低计算成本,并结合分层多分辨率模型,仅在几何结构至关重要的区域引入等变性处理,将使在适度计算资源条件下对百万原子体系进行全等变建模成为可能。与此同时,面向化学模拟优化的专用AI加速器所推动的软硬件协同设计,也将进一步加速这一转型。此外,超越基态化学,最具革命性的进展将来自多态MLIP,其能够同时描述激发态动力学、非绝热耦合效应、自旋–轨道相互作用以及相对论现象。
在可解释性方面,预计将取得实质性进展,从而缓解长期存在的“黑箱”问题。基于物理约束的可解释方法将发挥关键作用,例如利用符号回归从神经网络中提取人类可读的解析表达式,通过注意力可视化揭示驱动预测的关键分子特征,以及借助因果推断识别真正的化学因果关系。交互式可视化工具将帮助化学家实时探查模型的决策过程,弥合计算预测与化学直觉之间的鸿沟。
到2030时将出现完全自主的化学发现流水线:AI系统能够提出关于新化学现象的假设,设计验证这些假设的实验,利用自我改进的MLIP执行模拟,对结果进行解释,并生成新的科学认知。化学研究将日益由计算驱动,许多化学发现将首先在in silico中完成,再由实验加以验证。实时分子工程将使化学家能够以交互方式设计和优化分子,而AI则即时提供关于可行性、性质与合成路径的反馈。
在安全评估方面,预测型化学安全分析有望达到足够精度,从而在新化学品合成之前就影响政策制定;与此同时,AI智能体将使先进的化学模拟技术向更广泛的学科研究者开放。这些进展在经济层面也具有深远影响:更高效的药物设计将显著降低制药研发成本,计算设计的革命性能源技术有望加速落地,而强大模拟工具的普及将推动整个科研生态的发展。
参考链接:https://doi.org/10.1038/s43588-025-00930-6
文章改编转载自微信公众号:
原文链接:https://mp.weixin.qq.com/s/X95Uq8bJFv-0U-68MvGLAw | 





















