|
分子性质预测在生物制药领域的药物发现中至关重要,因为它有助于识别有前景的化合物,优化新疗法的开发效率。
2025年11月27日,清华大学王朝坤与李蓬团队在《Nature Communications》上发表论文,题为“Molecular Motif Learning as a pretraining objective for molecular property prediction”。
该研究提出一种无监督预训练方法MotiL,该方法可直接从原生分子图中学习分子表示,同时保留整体分子结构和基序级信息。该表示使MotiL在众多分子性质预测任务上,准确率超过了当前最先进的对比学习或预测式方法。
MotiL代码仓库:https://github.com/Young0222/MotiL
背景
对分子性质的精确预测能够显著加深对化学和生物系统的理解,从而推动药物发现、材料科学以及分子生物学的发展。小分子性质,例如毒性、血脑屏障通透性以及不良药物反应,对于药物的有效性和安全性至关重要。传统湿实验方法测定这些性质既耗时又昂贵,因此迫切需要开发可靠的计算模型,尤其是机器学习方法。此外,蛋白质在许多细胞功能中发挥关键作用,其折叠与稳定性等性质对于理解生物机制和设计治疗干预至关重要。
对于分子表示学习而言,生成能够准确反映底层化学结构与功能的表示至关重要。然而,现有方法由于在建模局部化学语义或层次子结构方面能力有限,难以有效捕获基序表示,例如小分子中的官能团或蛋白质中的氨基酸残基。例如,以往的对比学习方法往往依赖随机子图采样或全局级监督,这可能忽视在药理或结构上具有重要意义的基序。同样,预测式分子表示学习通过掩蔽子图、上下文或属性进行训练,也会破坏原始分子中的基序完整性,从而进一步妨碍关键化学性质的保留。
方法
MotiL总共包含三个阶段,分别为扩散预训练、双尺度训练、任务特定微调。这三个阶段分别实现全局结构感知、局部特征整合以及真实标签对齐。
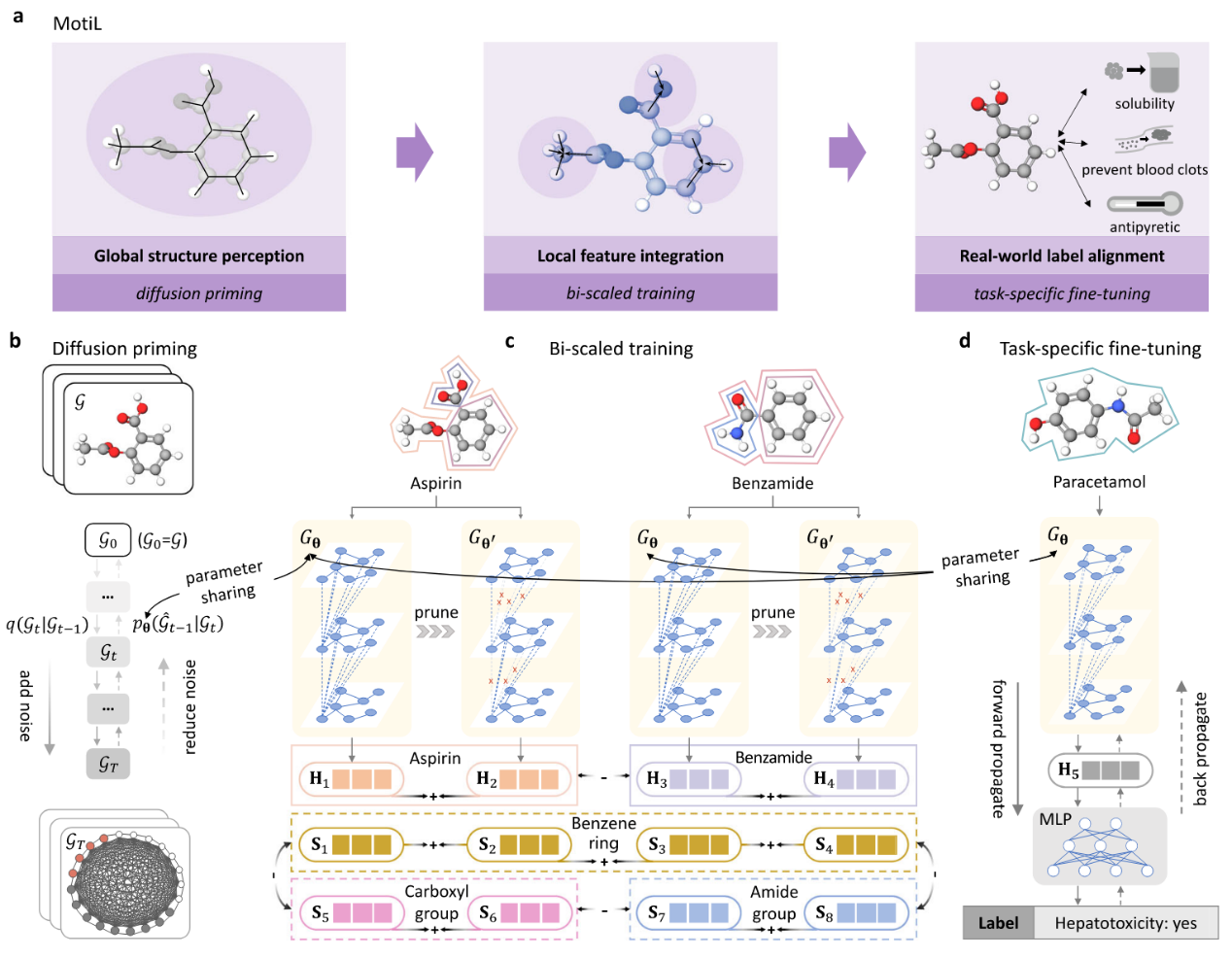
图1 MotiL工作流
在第一阶段,设计了分子扩散模型DiffMoM通过改变并纠正化学键对GNN参数进行预热。在第二阶段,基于原始网络和剪枝网络构建正样本对与负样本对,并设计了双尺度表示对齐机制,以确保来自同一图(同一分子)的表示保持一致,来自同一基序(即官能团)的表示保持一致。具体来说,在图层面,MotiL会为具有相同骨架的小分子以及具有相似三维结构和重叠化学功能的蛋白质生成相似表示;在基序层面,MotiL能够识别具有相同化学键的官能团,为其分配相似的基序表示。此外,MotiL还能在多种氨基酸中识别结构异构体并赋予相似表示,而对于结构差异较大的氨基酸则赋予不相似表示。在最后阶段,预训练后的GNN编码器为未见过的分子生成表示,随后将这些表示输入MLP分类器,以学习图表示与真实分子性质标签之间的关系。
结果
MotiL提升分子性质预测性能
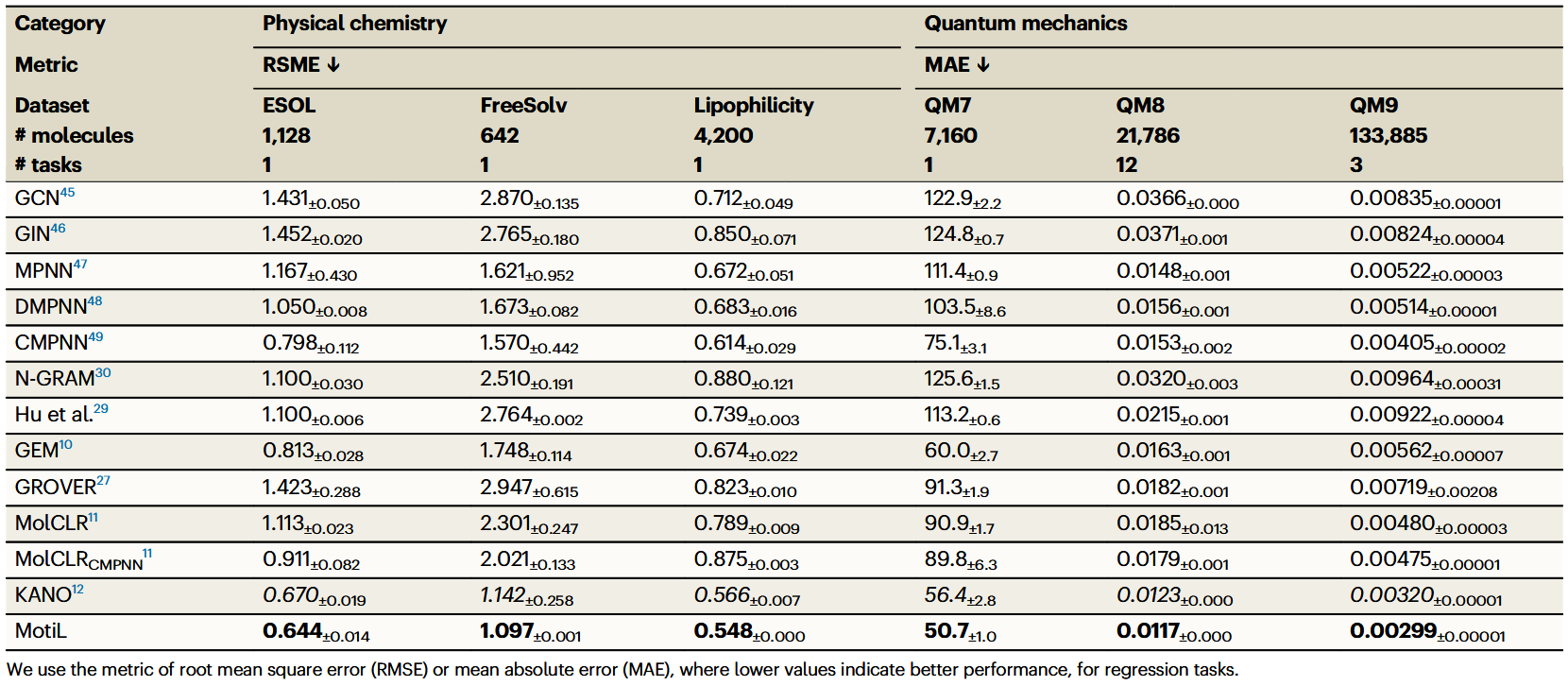
表1及表2展示了当前方法与MotiL在小分子性质预测的分类与回归任务上的实验性能。结果表明,MotiL在各数据集上均优于所有基线方法,相比次优方法平均相对提升3.0%。尽管KANO在表1和表2中表现较高,但它无法准确预测某些化疗药物或抗生素的血脑屏障通透性,以及部分分子的肝毒性。MotiL在特定案例中纠正了KANO的错误预测,并在整体准确率上超过KANO。基于数据增强的方法,如MolCLR、CMPNN和KANO虽然优于监督方法和预测式自监督方法,但它们往往忽视图增强对基序完整性的负面影响。相比之下,MotiL避免激进的数据增强,融合生成式扩散模型以提升鲁棒性,在各基准上实现持续更优的性能。这些结果凸显了基序保持型表示学习的有效性。
表1 生理学与生物物理领域小分子的性能比较
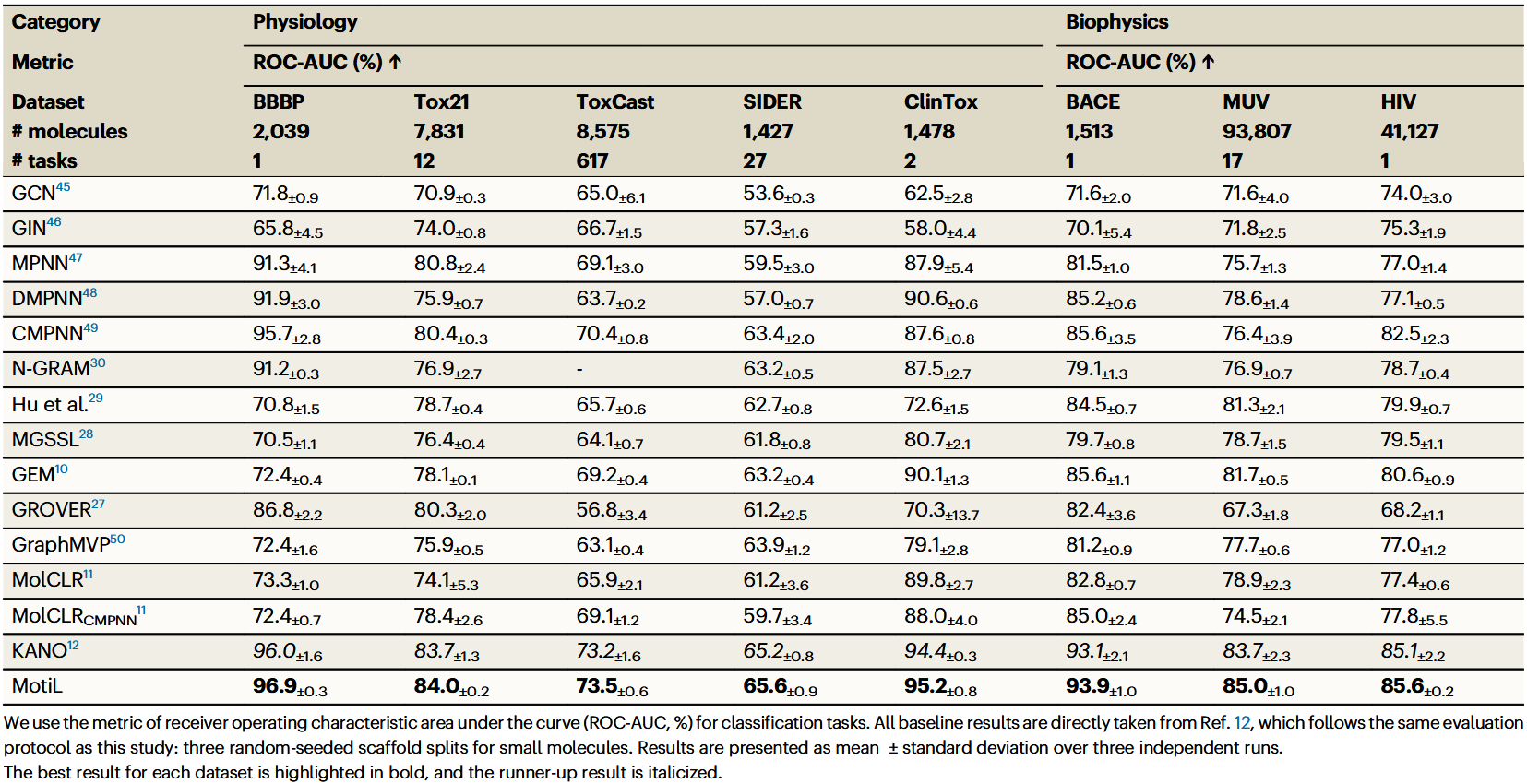
表2 物理化学与量子力学领域小分子的性能比较
接下来,在两个蛋白质数据集上评估不同方法的Fmax性能,结果见表3。结果表明,MotiL持续优于包括GearNet和CDConv在内的图方法,显示出其在计算生物学中的广泛应用潜力。
表3 蛋白质大分子的性能比较
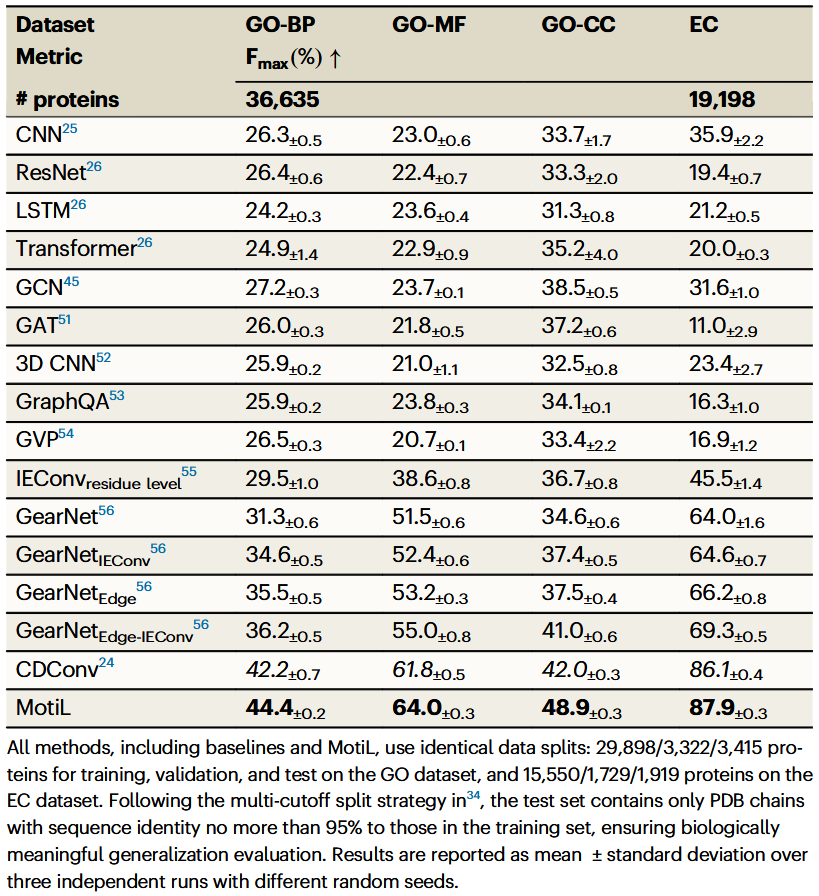
MotiL学习到与分子结构一致的小分子表示
使用t-SNE将MotiL学到的小分子表示投影到二维空间(图2a)。其中每个点代表一个分子,颜色表示该分子的骨架。Davies–Bouldin(DB)指数越低,表示簇内更紧密,簇间分离更明显。将MotiL与三种方法进行比较,分别为无预训练(不使用扩散预训练和双尺度训练)、带增强的分子对比学习MolCLR以及不带增强的分子对比学习(从MotiL中移除扩散预训练)。得到以下结论:无预训练效果最差,说明预训练对有效分子表示至关重要;带增强与不带增强的分子对比学习表现相近,合理的图增强有助于GNN区分分子但有时会产生误导,去除增强虽避免依赖,但忽视了对结构噪声鲁棒性的提升,因此并未优于带增强方法;MotiL在四个数据集上均实现清晰聚类。
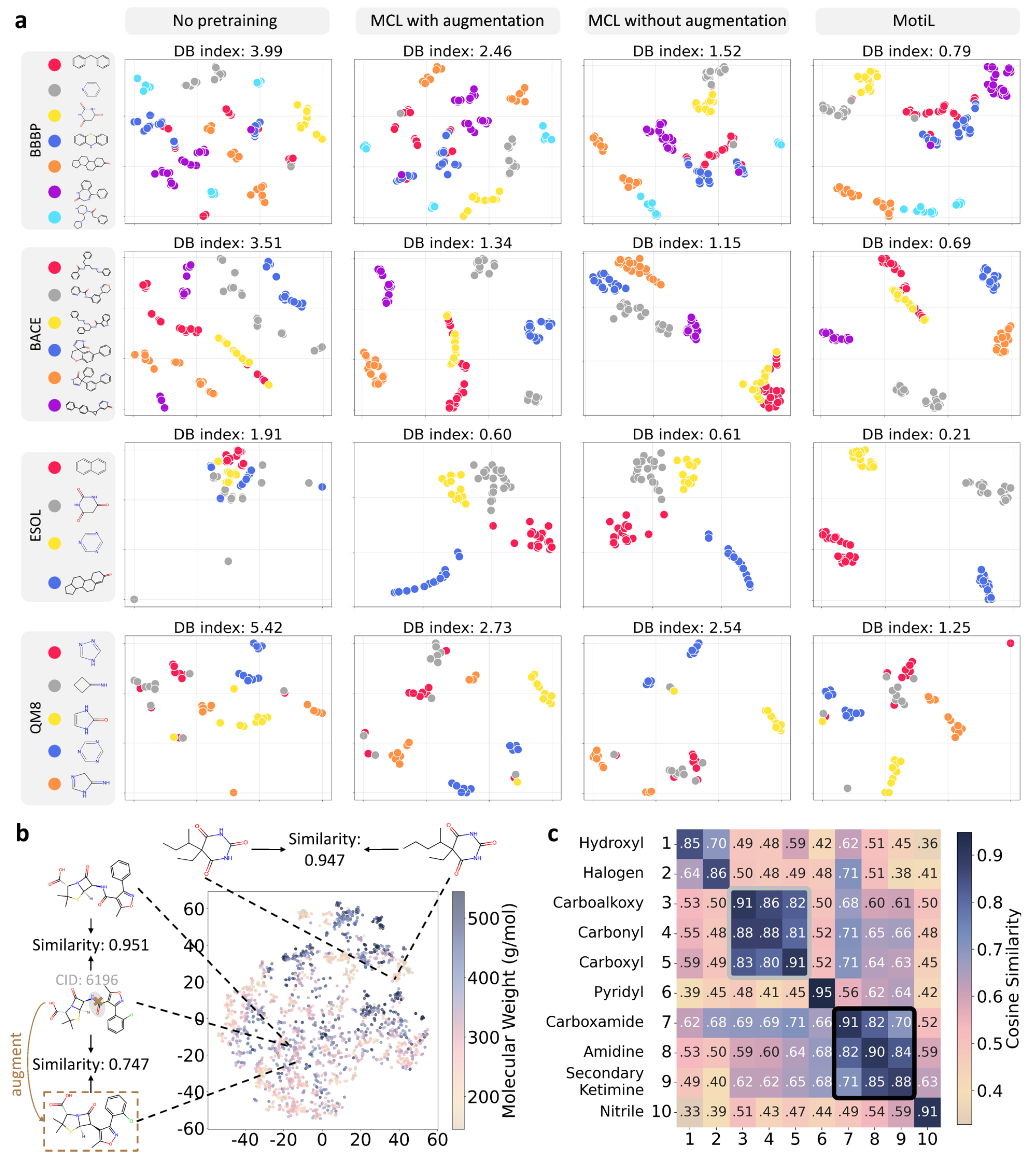
图2 MotiL学到的小分子表示分析
接着在多个数据集上可视化所有分子的t-SNE表示(图2b)。对于一对分子,通过RDKit指纹的Tanimoto系数计算其结构相似度(RDKFP相似度)。从图2b可见,RDKFP相似度高于90%的分子对在BBBP数据集的表示空间中通常彼此接近。此外,插入一个增强分子以验证图增强的影响。该增强分子通过对子图删除从CID为6196的分子生成(棕色虚线框标出)。由于被删除的酰胺基是关键官能团,增强分子与原分子的RDKFP相似度<75%,在表示空间中也被投影得更远。相比之下,与原分子相似度为0.951的另一个分子距离更近。值得注意的是,酰胺基的极性、形成氢键的能力以及增加分子刚性的潜力通常会降低分子通过血脑屏障的能力。这些结果表明,MotiL能同时捕获小分子的结构属性和功能属性并能区分由增强构造的易混淆分子对。
最后,通过计算余弦相似度分析10种常见官能团表示之间的关系。由图2c可知,相同官能团在不同编码器下仍保持相似表示,不同官能团的表示大多不相似。同时,一些官能团由于结构中原子和键的重叠而表现出较高相似度。上述分析表明,MotiL的双尺度学习机制能够使学到的基序表示与官能团的结构属性保持一致。
MotiL学习到与蛋白质结构和化学性质一致的大分子表示
使用UMAP将MotiL学习到的ATP结合蛋白表示投影到二维坐标空间(图3a)。随后应用基于密度的空间聚类算法DBSCAN识别并可视化各个蛋白的聚类ID。接下来,绘制了分子功能相关GO术语的Jaccard相似度热图,分析对象为包含10–40个蛋白的簇。图3b结果显示,簇内相似度普遍较高,簇间相似度相对较低。这表明,每个蛋白学习到的表示与其分子功能保持一致。随后,聚焦具体案例考察蛋白质的三维结构。图3c结果显示,在第3簇中,蛋白4AU8-A、1G3N-A、2BPM-A、5XVU-A和3VN9-A在结构上彼此高度相似。此外,这些同簇蛋白还共享GO注释,例如蛋白激酶活性。最后,计算了20种氨基酸表示之间的相似度,并以热图形式展示。由图3d可知,相同氨基酸在不同编码器下表示高度相似,不同氨基酸之间相似度较低。此外,MotiL能够准确识别具有相似芳香环结构的分子(高度相似),结构异构体(相似),侧链结构差异较大的分子(不相似)。
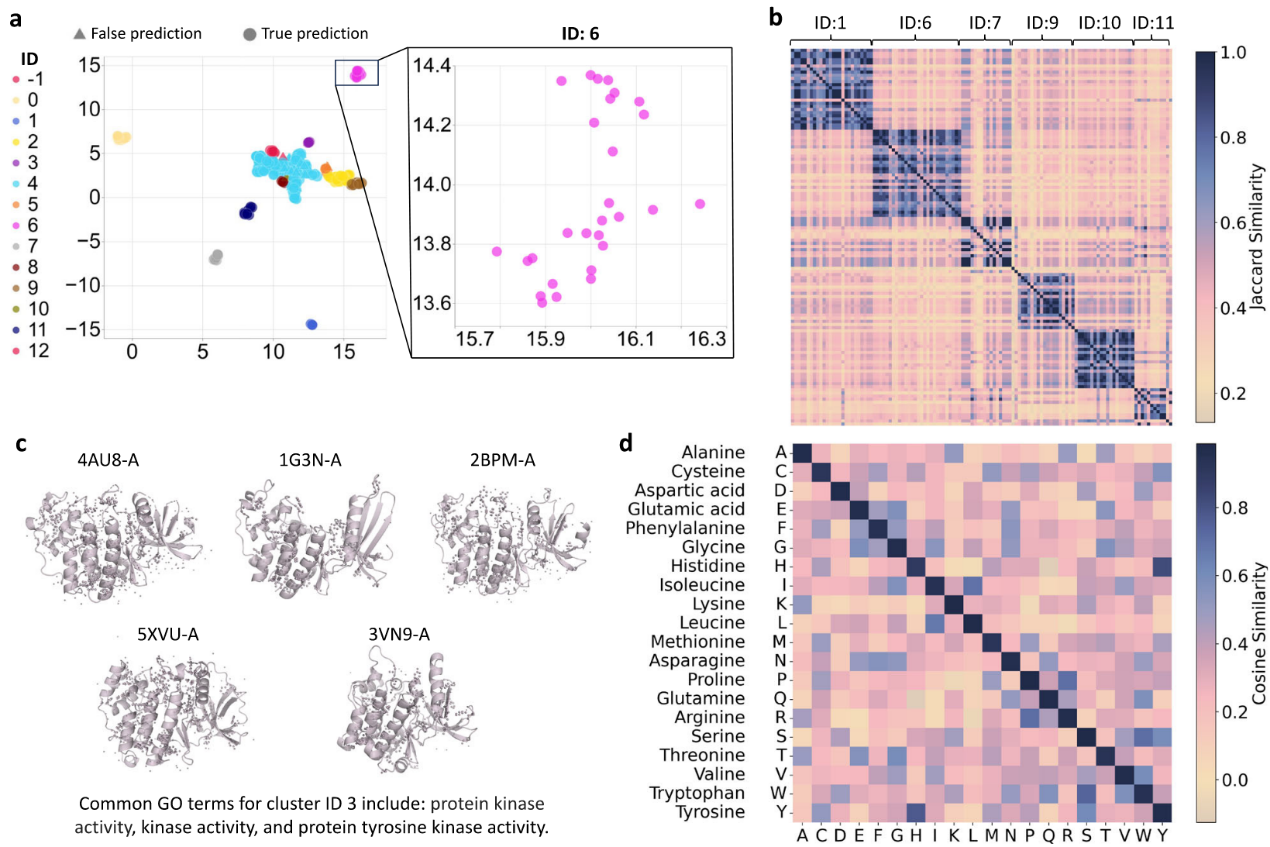
图3 MotiL学到的大分子表示分析
MotiL纠正最强基线模型的错误预测
在两个与毒性相关的基准数据集BBBP与SIDER上,对MotiL与最强基线KANO的预测分歧进行了系统评估。图4a展示了完整数据集上的分歧统计,图4b给出了活性悬崖实验结果。该实验用于评估模型在“结构高度相似但标签不同”的困难分子对上的鲁棒性。这些结果表明,MotiL在处理模糊或困难样本时优于KANO。图4c-d分别展示了BBBP与SIDER中的代表性示例。这些定性结果补充了统计分析,并揭示了MotiL在化学结构复杂或临界样本中的判别机制。
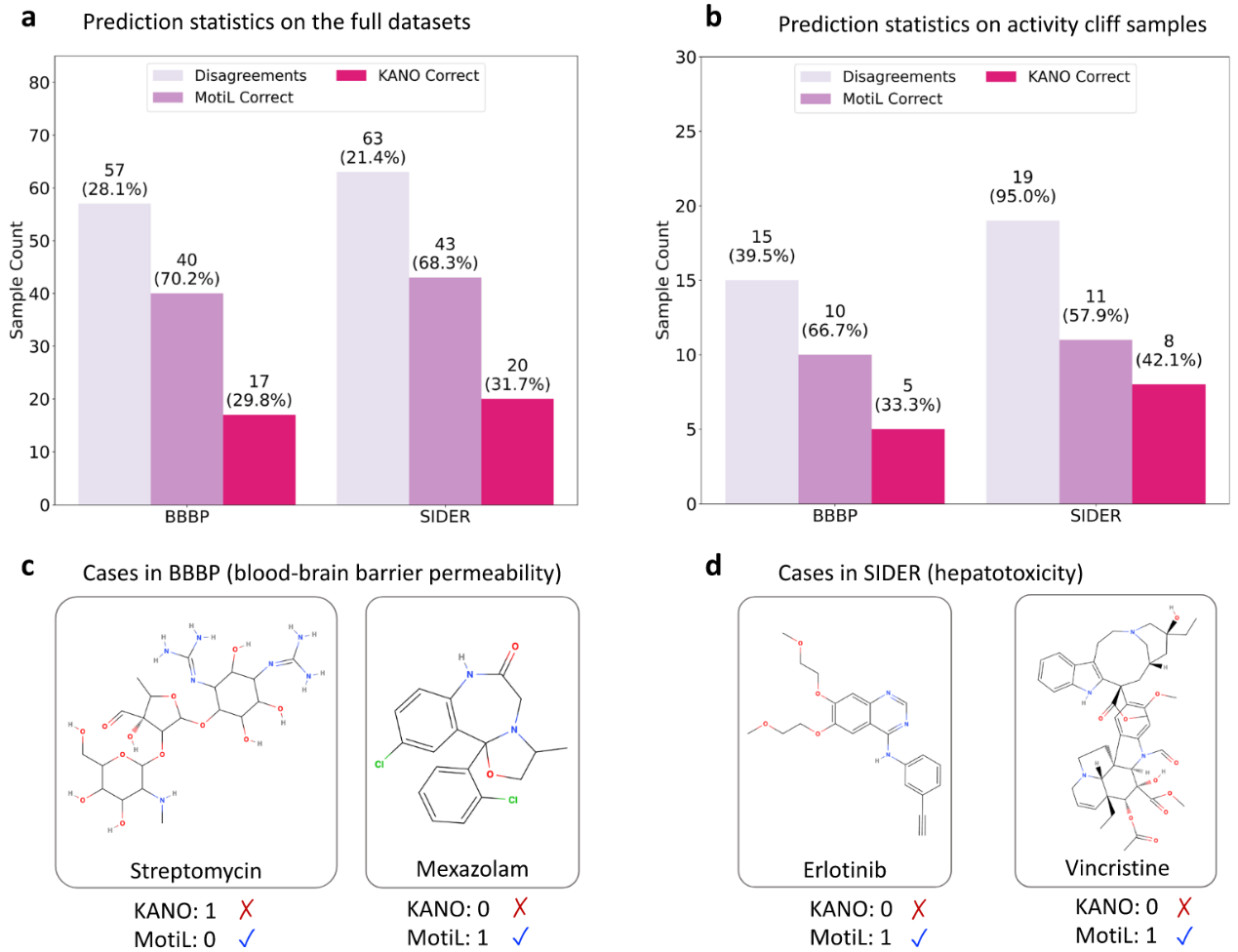
图4 KANO与MotiL在BBBP和SIDER上预测差异的定量与定性分析
总结
本文提出的分子预训练表示学习方法将生成式扩散模型、网络剪枝和双尺度训练损失集成到MotiL中,以实现分子基序学习。实验结果表明,MotiL在14个小分子基准与2个蛋白质大分子基准上均取得了优越性能。未来仍有多个值得探索的方向,例如在扩散预训练阶段探索更先进的生成模型,可能进一步提升分子表示质量;探索生成式扩散模型与表示学习在新分子设计中的混合应用。分子设计能力的提升可能带来更有效的治疗方案,更深入的生物过程理解以及合成生物学与疾病诊断的新机遇。
参考链接:https://doi.org/10.1038/s41467-025-66685-w
文章改编转载自微信公众号:智药邦
原文链接:https://mp.weixin.qq.com/s/MNeDiztHi6fQ4gQ6m9q-8A | 





















