理解蛋白质–配体相互作用是生命科学诸多领域的基础:从阐明细胞过程到开发疾病新疗法,都离不开对其机制的描述。这类相互作用能够触发信号级联反应、催化化学反应,或调控基因表达等关键生物学事件。作为细胞的“主力军”,蛋白质往往通过与小分子配体或其他蛋白质发生特异性结合来实现功能。然而,蛋白质结构具有天然的动态性,配体结合也常诱导其发生构象变化;因此,获得准确的结合构象是基于结构的药物设计的重要前提。通过在分子层面更精确地理解这些相互作用,我们可以更深入认识生物系统如何运转,以及更有针对性地调控其功能,从而实现治疗目的。
近日,中国科学院上海药物研究所郑明月团队提出蛋白–配体复合物柔性结构建模新方法PackDock。该方法将生成式AI与物理算法相结合,用于预测柔性蛋白–配体对接构象,并在多种应用场景中展现出良好的精度与效率,同时具备较强的泛化能力。相关研究成果以“Flexible protein–ligand docking with diffusion-based side-chain packing”为题,于2025年12月24日在Proceedings of the National Academy of Sciences of the United States of America(美国国家科学院院刊)在线发表。

背景
蛋白质的结构对于基础生物学和药物设计至关重要。过去几十年里,基于结构的药物设计(SBDD)已成为开发新药的核心策略;随着结构生物学技术与从头蛋白质折叠算法的快速发展,获取蛋白质三维结构变得前所未有地便捷。然而,蛋白质结构本质上是动态的,配体结合常伴随结合口袋内的构象变化,而现有结构多以“静态快照”的形式呈现,难以满足SBDD的复杂需求:例如,未结合配体的apo结构或结合非同源配体的holo结构,都可能导致对新配体真实结合模式的错误假设。因此,尽管准确的结合构象是SBDD的关键前提,它在实际研究与应用中仍往往难以直接获得。
传统分子对接算法通常依赖迭代式的构象采样与打分评估。为保证计算效率,这类方法往往忽略蛋白质柔性,从而缩小搜索空间,但这可能错过关键的口袋构象变化并导致错误的结合模式。近年来,深度学习方法通过回归或生成式建模在一定程度上能够跳过反复的“采样—评估”流程,显著提升对接速度,并在部分场景下带来更高精度;但由于神经网络对物理约束的显式建模不足,仍可能产生几何或能量上不合理的构象,从而对结果的可解释性与可信度提出挑战。与此同时,共折叠(Co-folding)方法的兴起进一步改变了关于蛋白质–配体复合物结构预测的讨论:相较于传统对接方法,它们在多个任务上展现出更强的性能,但多项基准研究也提示,现有共折叠框架尚未学习到真实的物理相互作用规律。
在这项工作中,研究团队提出了柔性蛋白质建模框架PackDock,通过整合物理建模与深度学习方法来表征蛋白质–配体相互作用。其核心模块PackPocket结合等变图神经网络与扩散模型建模结合口袋内侧链构象的多峰分布,从蛋白质“自由态”与“配体结合态”的构象分布中采样,并通过预测侧链扭转角实现对蛋白质柔性的高效建模。为系统评估该方法,研究团队设计了一系列面向真实药物设计需求的基准实验,包括侧链恢复、柔性重对接,以及在apo、holo与预测折叠结构上的交叉对接测试,从而全面评估PackDock在不同结构来源下的柔性对接性能。此外,团队还在一个面向ALDH1B1的真实药物发现项目中,成功识别出具有新型骨架、纳摩尔级亲和力的候选化合物,进一步验证了PackDock的实用价值。
结果
1 PackDock架构
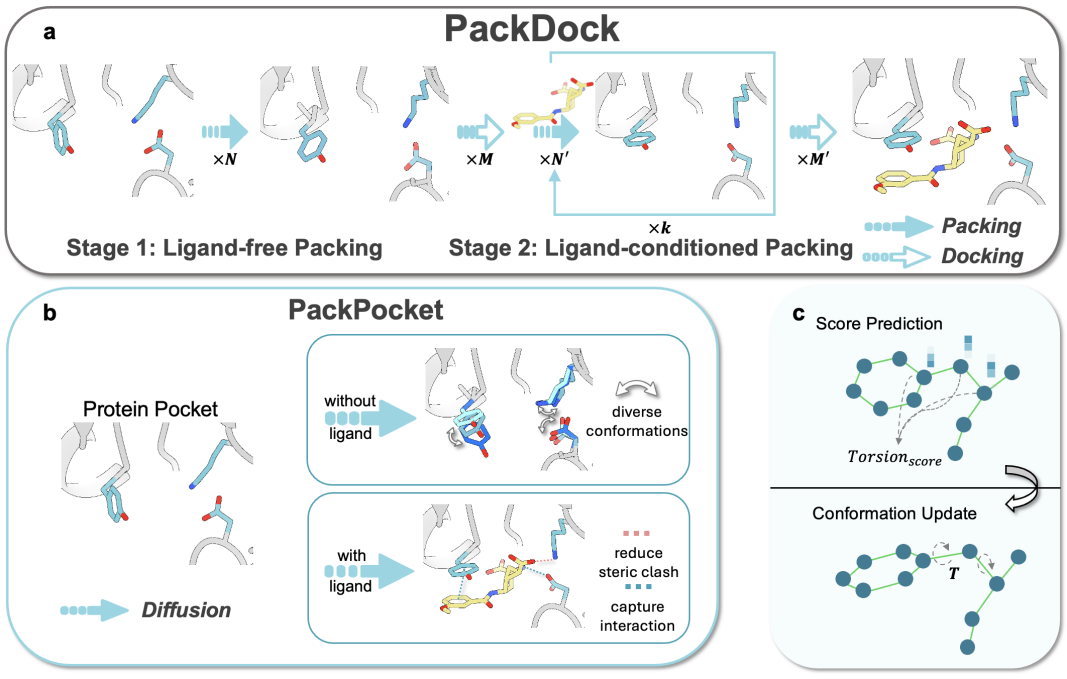
图1.PackDock方法示意图。a:PackDock工作流程;b:PackPocket对侧链构象分布的建模;c:等变图神经网络预测侧链扭转角。
如图1所示,PackDock同时考虑蛋白质在无配体(自由态)与配体结合态下的构象分布,从而更精准地建模柔性蛋白–配体复合物的结合构象。具体而言,核心模块PackPocket结合等变图神经网络与生成式建模策略,学习侧链构象空间的能量景观,并在不同状态分布中采样口袋侧链构象,以描述结合过程中可能发生的构象变化。此外,PackDock框架可兼容多种刚性对接算法,从而进一步提升柔性对接性能。
2 柔性对接的基础:蛋白侧链构象预测的准确性
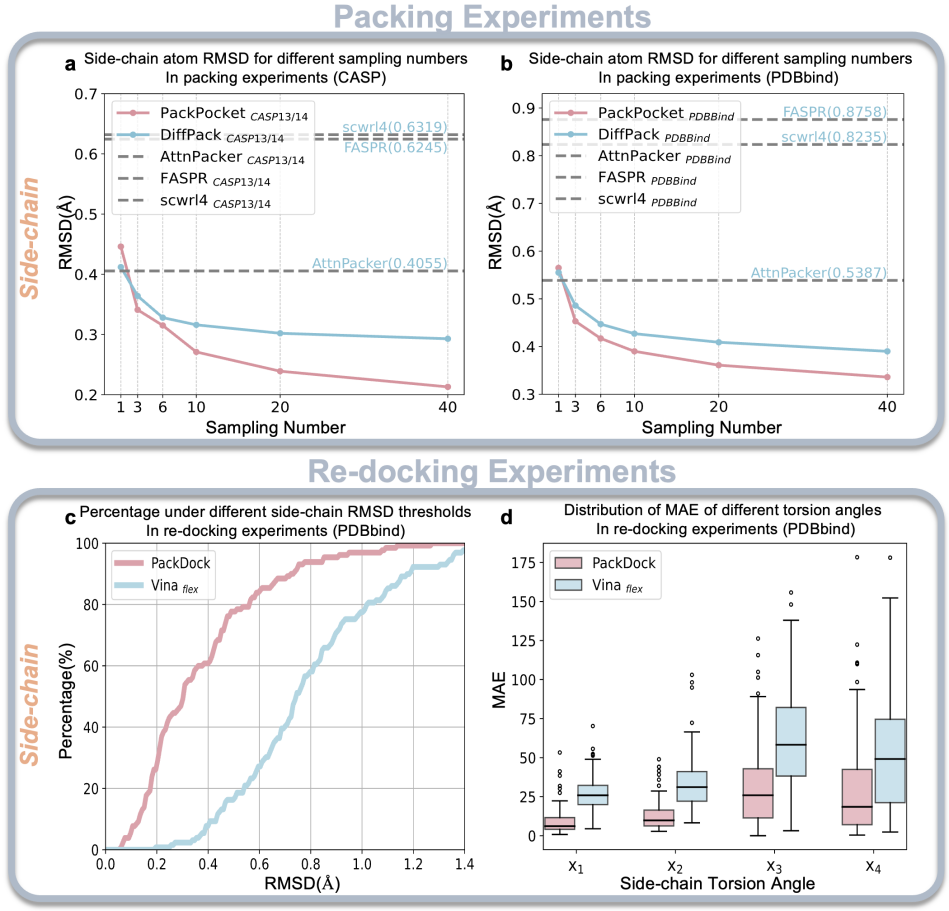
图2. PackDock与其他方法的侧链预测准确度对比。a:无配体条件下的Packing性能;b:以配体为条件的Packing性能;c:柔性重对接过程中的侧链原子RMSD;d:柔性重对接过程中的侧链扭转角MAE。
蛋白质构象并非一成不变,尤其在配体结合口袋区域,口袋“呼吸”或变构效应常会驱动侧链重排,从而形成多种可能的构象状态。然而,现有方法多侧重于评估单一结构的准确性,难以生成多样且具有代表性的口袋构象集合。为此,PackDock的核心模块PackPocket采用生成式建模学习并采样口袋侧链构象分布,并可进一步引入配体条件信息,以提升侧链预测的准确度(图2)。此外,侧链构象空间极其庞大,传统柔性对接往往需要在“采样—评估”的迭代过程中耗费大量计算以探索该空间;相比之下,PackPocket通过直接建模与采样侧链构象分布,避免了大量迭代计算步骤,从而在提升精度的同时显著提高了计算效率。
3 真实结构偏差下的鲁棒性:apo与holo结构的交叉对接
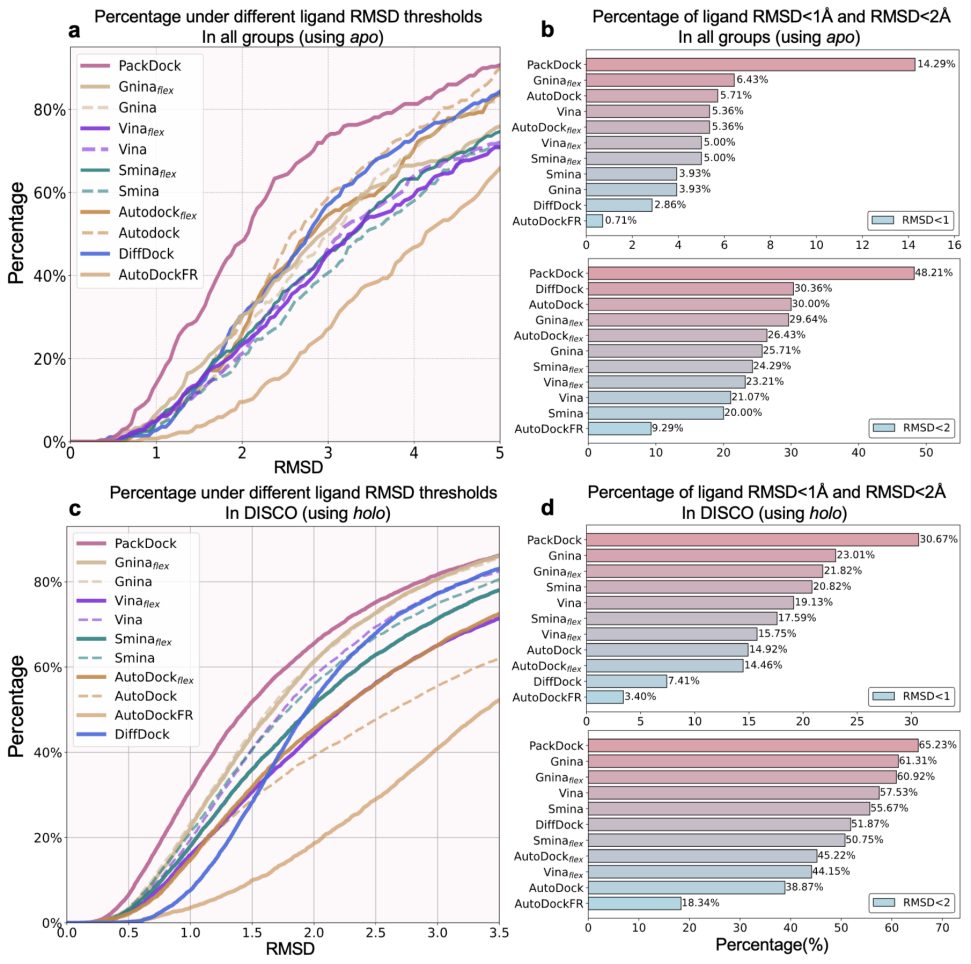
图3.PackDock与其他方法的柔性对接性能对比。a:使用apo结构进行对接的性能;b:使用apo结构对接时RMSD<1Å和<2Å的比例;c:使用holo结构进行对接的性能;d:使用holo结构对接时RMSD<1Å和<2Å的比例。
在大多数实际应用中,获得与目标配体结合的holo结构往往十分困难,通常只能依赖apo结构或非同源配体的holo结构来开展分子对接研究。在这类场景下,若将蛋白质视为刚性结构、仅考虑配体柔性,往往导致错误的相互作用模式,从而限制对接结果的可靠性。为贴近真实应用,研究者分别以apo与非同源holo结构模拟上述输入条件并进行评测。如图3所示,PackDock在以apo或非同源holo作为输入时的柔性对接性能均显著优于现有方法,体现了其在真实结构偏差条件下的鲁棒性与实用价值。
4 预测结构的可用性:折叠结构模型上的交叉对接
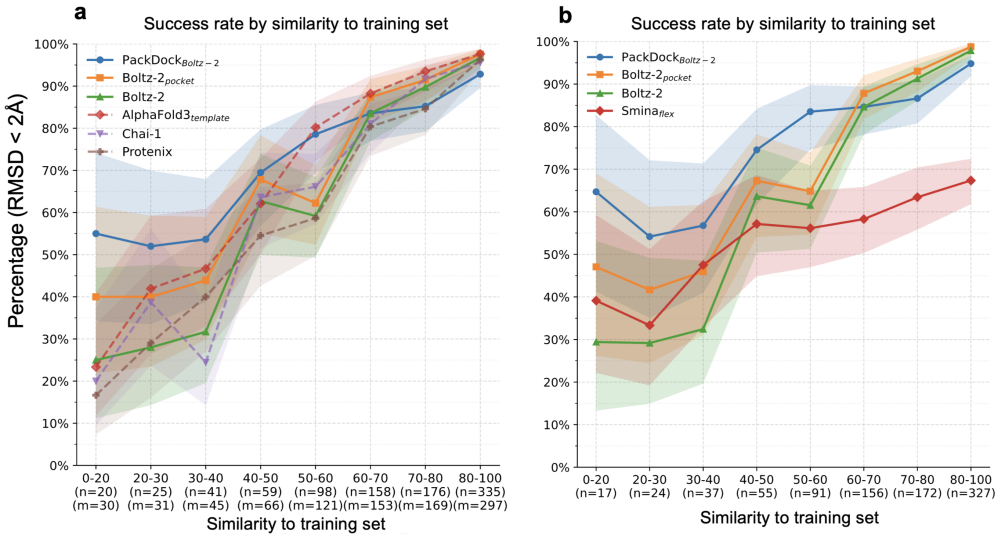
图4. 以共折叠方法预测结构作为输入时PackDock的对接性能。a:与其他共折叠方法在不同训练集相似度测试集上的对接性能对比;b:在主链RMSD<2Å的测试数据上的对接性能。
过去几十年,实验结构生物学已解析约10万个蛋白质结构,但这仍仅覆盖已知蛋白质序列空间的一小部分,因此人们寄希望于借助蛋白质折叠算法探索更广阔的序列图景。近年来,共折叠方法在预测过程中显式引入配体信息,相比传统分子对接在多个任务上展现出显著优势,也为SBDD带来了令人兴奋的新机遇。然而,多项基准测试同样揭示了其局限:部分方法可能尚未学习到真实的物理相互作用规律,而更依赖对训练集中配体姿态的“记忆”,从而在分布外结构上出现明显的性能衰减,引发了对其在药物发现应用中泛化能力的担忧。
PackDock采用混合策略,将基于物理的分子对接与深度学习驱动的受体柔性建模相结合;这种组合在‘记忆式泛化’失效时尤为有效。为评估其泛化性,研究者使用Runs N Poses数据集,并将评估范围限定在Boltz-2训练截止日期(2023年6月)之后发布的结构,同时依据测试样本相对于训练集的相似度(SuCOS)进行分层分析。
如图4所示,PackDock在低相似度(<60%)条件下取得了显著更高的对接成功率。尤其值得注意的是,当相似度低于40%时,PackDock的性能相比Boltz-2提高约20%,相比Boltz-2-pocket提高约10%。这类低相似度样本往往最具挑战性,却也更贴近未来药物发现中常见的“分布外”应用场景。在高相似度条件下,当共折叠方法的准确率接近完美(>90%)时,PackDock的表现略低,但整体仍具有竞争力。总体而言,共折叠方法代表了SBDD的重要发展方向,但在物理可解释性与泛化性方面仍面临根本挑战;因此,与其将其视为传统对接的替代方案,不如将二者的优势互补结合,以覆盖更广泛、更真实的应用场景。
5 应用验证:前瞻性虚拟筛选实验
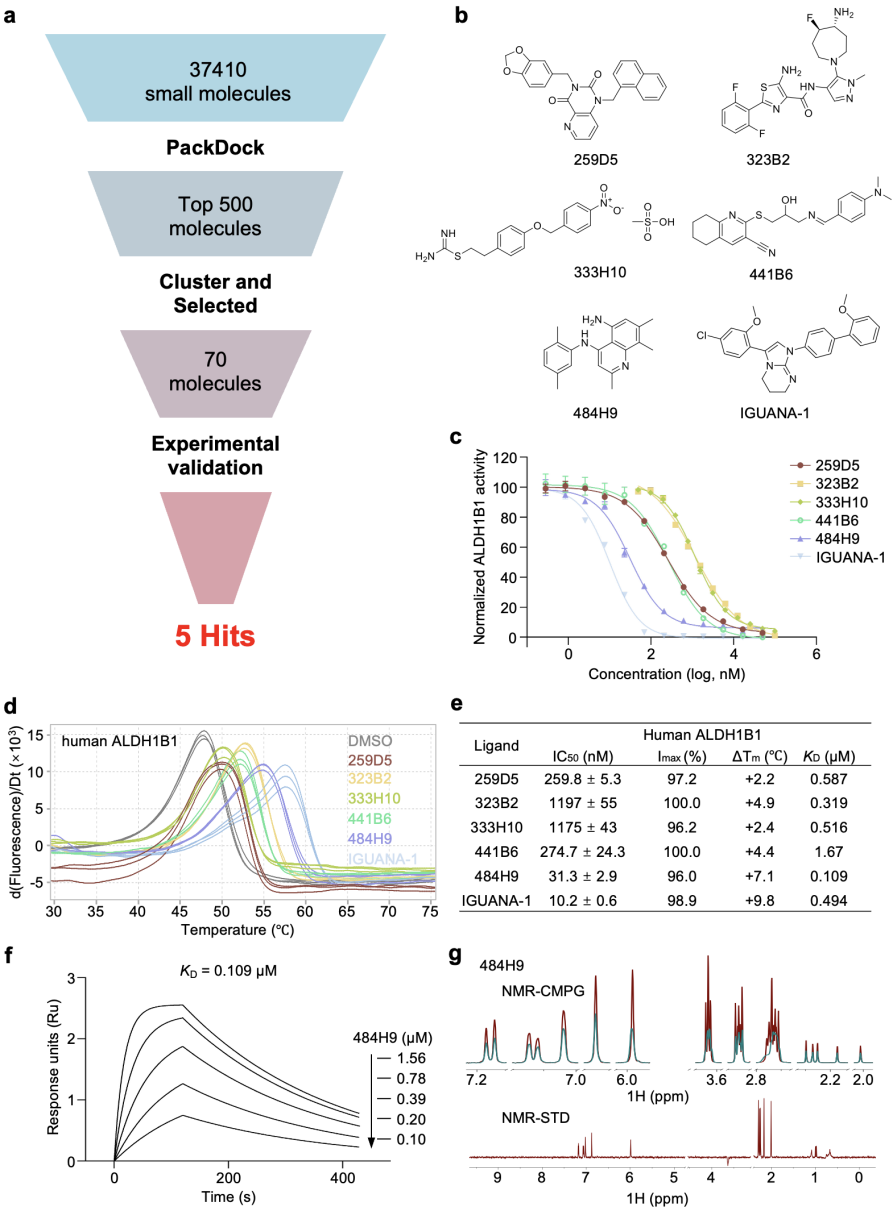
图5. PackDock筛选ALDH1B1的新骨架化合物。a:筛选流程示意图;b:筛选得到的hit分子与参考分子IGUANA-1的化学结构;c:5个hit分子及参考分子的剂量–反应曲线;d:hit分子与参考分子对ALDH1B1蛋白热稳定性的影响;e:实验活性鉴定结果汇总;f:SPR测定484H9与ALDH1B1的结合亲和力;g:484H9与ALDH1B1的核磁共振实验。
为了评估PackDock在实际药物发现流程中的应用能力,研究团队以ALDH1B1为靶点开展了一项前瞻性虚拟筛选实验,如图5所示。
通过使用PackDock对内部小分子库进行筛选,并对优选化合物进行多层级实验验证,最终获得5个具有新型骨架的ALDH1B1抑制剂,其中1个化合物表现出纳摩尔级的结合亲和力。该结果表明,PackDock不仅在基准任务上具备优势,也能够在真实筛选中产出具有实际价值的命中分子,体现了其面向药物发现应用的潜力。
讨论
结构生物学技术与基于人工智能的蛋白质折叠算法迅速发展,使我们能够以前所未有的速度获得更多新型生物靶标的三维结构。然而,这些结构往往以“静态快照”的形式呈现,难以全面反映蛋白质在细胞环境中的动态特性,从而限制了我们对蛋白质–配体相互作用的深入理解。为应对这一挑战,研究团队提出了PackDock框架,旨在弥补静态结构在描绘结合过程与受体柔性方面的不足。
PackDock将基于物理的分子对接与深度学习驱动的受体柔性建模相结合。研究者通过多项系统性评测验证了其能力:
1)侧链packing与柔性重对接实验表明PackDock能够准确预测结合位点侧链构象;
2)在以apo结构、非同源holo结构以及共折叠预测结构为输入的交叉对接测试中,PackDock展现出在真实结构输入条件下柔性对接的实用价值;
3)在针对ALDH1B1的前瞻性虚拟筛选中,PackDock成功识别出具有新型骨架且达到纳摩尔级亲和力的抑制剂,进一步证明其在实际药物发现中的应用潜力。
总体而言,PackDock能够适配多来源的蛋白结构输入,并以较高精度预测蛋白–配体复合物构象,从而推动对生物系统中蛋白–配体相互作用的机制理解。作者认为,与其期待深度学习完全替代传统经验或物理方法,更值得关注的是如何整合不同方法的优势,以更有效地解决实际科学问题。
上海药物研究所硕士研究生张润泽、上海药物研究所博士研究生江欣雨、浙江大学与上海药物研究所联合培养博士研究生曹端华,以及上海药物研究所硕士研究生王照坤为本文共同第一作者。上海药物研究所郑明月研究员为本文通讯作者。本研究获得国家自然科学基金、国家重点研发计划、临港实验室、中国科学院战略性先导科技专项等资助。
原文链接
R. Zhang,X. Jiang,D. Cao, et al. Flexible protein–ligand docking with diffusion-based side-chain packing, Proc. Natl. Acad. Sci. U.S.A. 122 (52) e2511925122 (2025).
https://doi.org/10.1073/pnas.2511925122
本文转自【人工智能药物设计】公众号
文章改编转载自微信公众号:智药邦
原文链接:https://mp.weixin.qq.com/s/Da3kBpI0PhBf-laLSG7DLg?scene=1&click_id=65 | 





















