本文解读 Nature Computational Science 2026 年论文《De novo design of functional nucleic acids of aptamers》,该研究提出融合核酸大语言模型(NA-LLMs)与高通量 SELEX 的 InstructNA 框架,无需依赖三维结构即可从头设计功能核酸(FNA)。针对 LOX1 和 CXCL5 蛋白靶点,其生成的强结合适配体数量较传统 HT-SELEX 分别提升 100% 和 200%,部分适配体与原始序列相似度低至 38%,同时保持高结合亲和力(最低 KD 达 6.6 nM),为分子识别、基因调控等领域提供高效设计工具。

在分子生物学的工具箱里,功能核酸(FNA)是一类 “多才多艺” 的明星分子 —— 它们不仅能存储遗传信息,还能像抗体一样特异性识别目标分子(适配体)、像酶一样催化化学反应(核酶),甚至能调控基因表达,在临床诊断、药物递送、生物传感等领域具有不可替代的价值。但长期以来,功能核酸的从头设计一直是科研界的 “硬骨头”:核苷酸序列空间极其庞大,传统实验筛选方法成本高、效率低,而计算设计又受限于核酸结构的高灵活性,难以建立稳定的序列 - 功能关系。
直到这篇发表在Nature Computational Science的研究问世,科学家们用 “核酸大语言模型 + 高通量筛选” 的组合拳,打造出名为InstructNA的功能核酸设计框架,彻底改变了功能核酸的设计范式。它无需依赖三维结构信息,就能快速生成高活性、高多样性的功能核酸,让 “按需设计” 成为现实。
一、功能核酸设计:长期困扰科研界的 “双重难题”
功能核酸(FNA)是一类具有特殊生物功能的 DNA 或 RNA 分子,涵盖适配体、核酶 / 脱氧核酶、基因调控元件等多种类型,凭借其独特的分子识别、催化和调控能力,在多个领域展现出广阔应用前景。然而,功能核酸的设计始终面临两大核心挑战:一方面,核苷酸序列空间极为庞大,一个仅 30 个核苷酸组成的核酸分子,可能的序列组合就高达 4³⁰种,传统实验筛选方法根本无法全面覆盖;另一方面,核酸结构具有极高的灵活性,序列与功能之间的映射关系复杂且难以捉摸,缺乏像蛋白质那样成熟的序列 - 结构 - 功能设计规则。
传统的 SELEX 筛选方法虽然是功能核酸发现的主流手段,但存在诸多固有缺陷:不仅成本高昂、周期漫长,还容易受到 PCR 偏差的影响,导致筛选结果偏向扩增效率高的序列,而非功能最优的序列。而现有计算设计方法也存在明显短板,多数方法依赖精准的核酸三维结构,而实验测定的核酸结构数量远少于蛋白质;同时,现有生成模型多依赖目标特异性的小规模 SELEX 数据,难以学习全面的序列 - 功能关系,泛化能力有限,难以适配多种类型的功能核酸设计需求。
二、AI 破局:InstructNA 的全能设计框架
2.1 框架核心逻辑:大语言模型 + 高通量筛选的双向赋能
InstructNA 的核心创新在于将核酸大语言模型(NA-LLMs)的语义理解能力与高通量 SELEX(HT-SELEX)的实验数据优势相结合,构建闭环设计流程,具体分为五步:
数据准备:收集目标功能核酸的 HT-SELEX 实验序列,构建高质量训练集;
模型适配:用 SELEX 数据对预训练核酸大语言模型持续预训练,打造领域适配的 FNA-LLM;
解码器训练:在 FNA-LLM 基础上训练轻量级解码器,实现从 latent 空间到核酸序列的精准解码;
迭代优化:通过自研 HC-HEBO 算法,在 latent 空间进行功能导向的定向进化;
实验验证:验证生成序列功能,将结果反馈模型完成迭代。
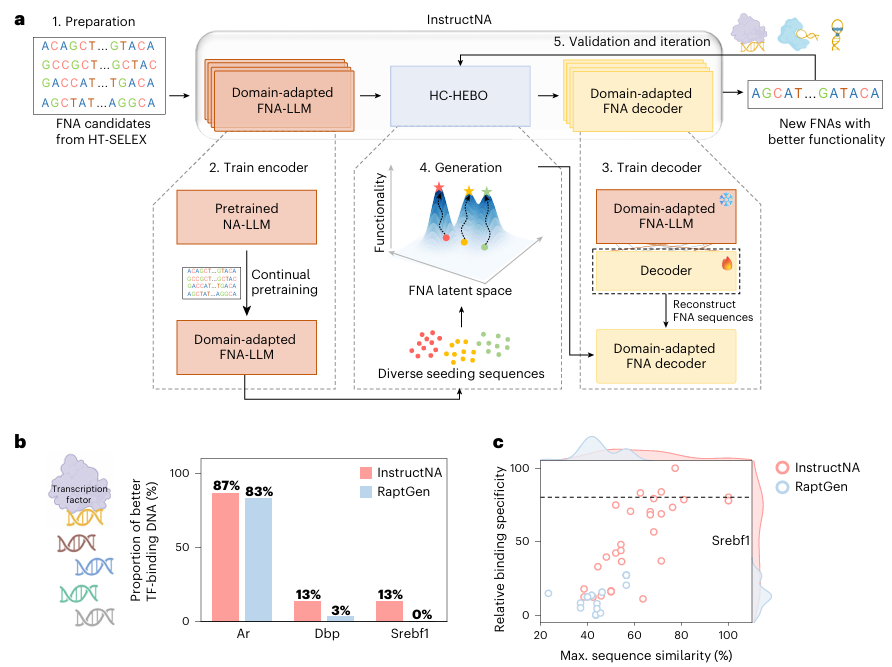
图一 InstructNA功能核酸从头设计框架
这套框架无需三维结构,仅通过序列数据即可学习功能核酸的语义特征与序列 - 功能关系,适配多种类型功能核酸设计。
2.2 关键技术:让 AI “看懂” 核酸的功能密码
语义增强的序列表示:持续预训练让模型捕捉核酸深层语义,latent 空间与真实序列空间的相关性显著优于传统模型;
HC-HEBO 优化算法:融合爬山法(HC)与异方差进化贝叶斯优化(HEBO),限制搜索空间的同时实现高效优化,平衡序列功能与多样性;
多源种子序列策略:融合高频率序列、聚类中心序列和高特异性序列作为初始种子,提升生成序列质量与多样性。
2.3 量化功能与序列特性
InstructNA 通过关键公式量化核酸功能与序列特征,为设计优化提供依据:
结合特异性评分(评估转录因子结合 DNA 特异性):
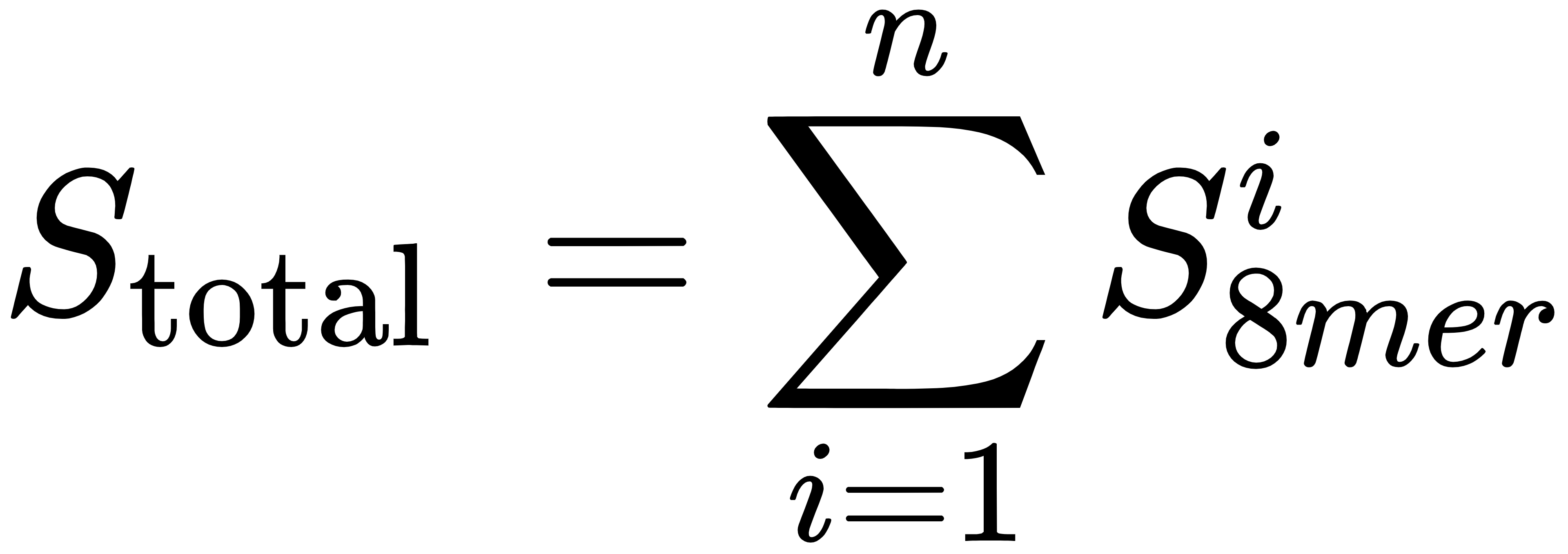
为第 i 个 8-mer 基序的蛋白质结合微阵列荧光强度,n 为序列中 8-mer 基序总数。
归一化相对结合特异性(便于不同序列比较):
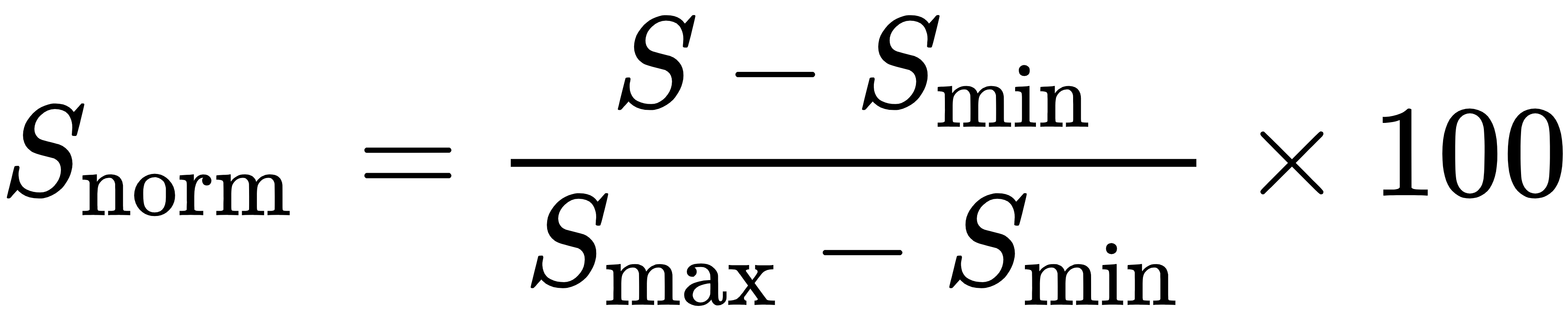
其中Smin和Smax分别为数据集中最小和最大结合特异性评分。
三、关键结果:功能与多样性的双重突破
3.1 转录因子结合 DNA:特异性与多样性双优
语义捕捉更精准:latent 空间与真实序列空间的皮尔逊相关系数显著高于 DNABERT、RaptGen 等基线模型;
分类性能更优异:在 AUROC、F1 值、准确率等多项指标上优于现有模型,序列 - 功能关系学习能力更强;
生成质量更高:针对 Ar、Dbp、Srebf1 靶点,分别有 87%、13%、13% 的生成序列结合特异性高于原始 HT-SELEX 高频序列,部分序列相似度低于 80%,多样性突出。
3.2 蛋白结合适配体:活性提升,突破筛选瓶颈
强结合适配体数量翻倍:LOX1 靶点从 2 个增至 4 个,CXCL5 靶点从 1 个增至 3 个,数量分别提升 100% 和 200%;
结合亲和力更优:LOX1 靶点 G1L(KD=12.9 nM)、CXCL5 靶点 G1C(KD=6.6 nM),均优于原始 HT-SELEX 最优序列;
序列多样性极高:LOX1 靶点 G1L 与原始最优序列 T1L 相似度仅 38%,三维结构更复杂,通过更多氢键结合靶蛋白。
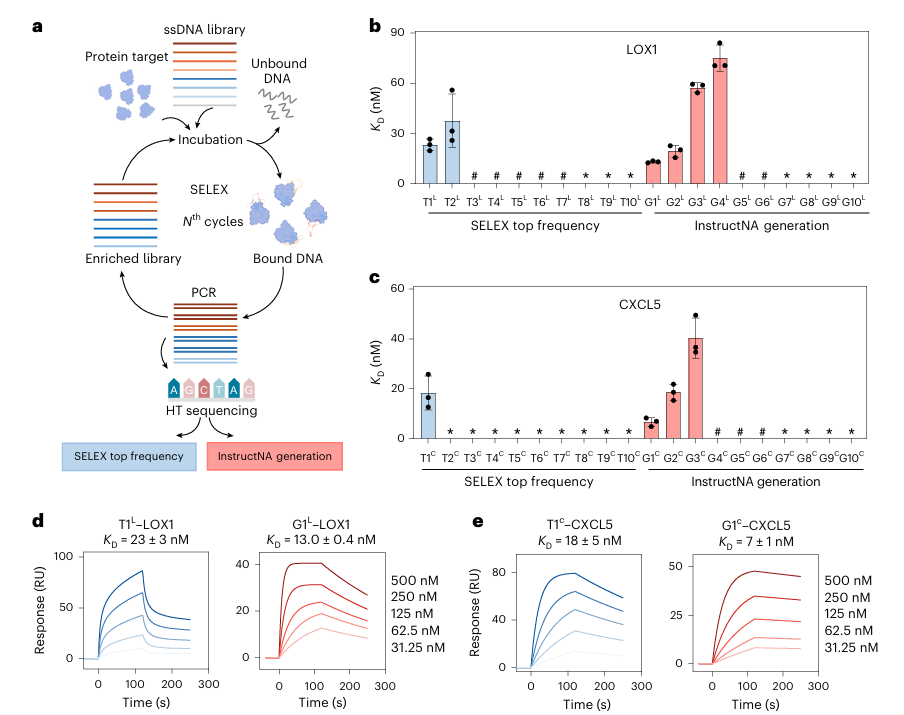
图二 InstructNA生成适配体的结合活性验证
3.3 模型鲁棒性与泛化性:适配多场景设计
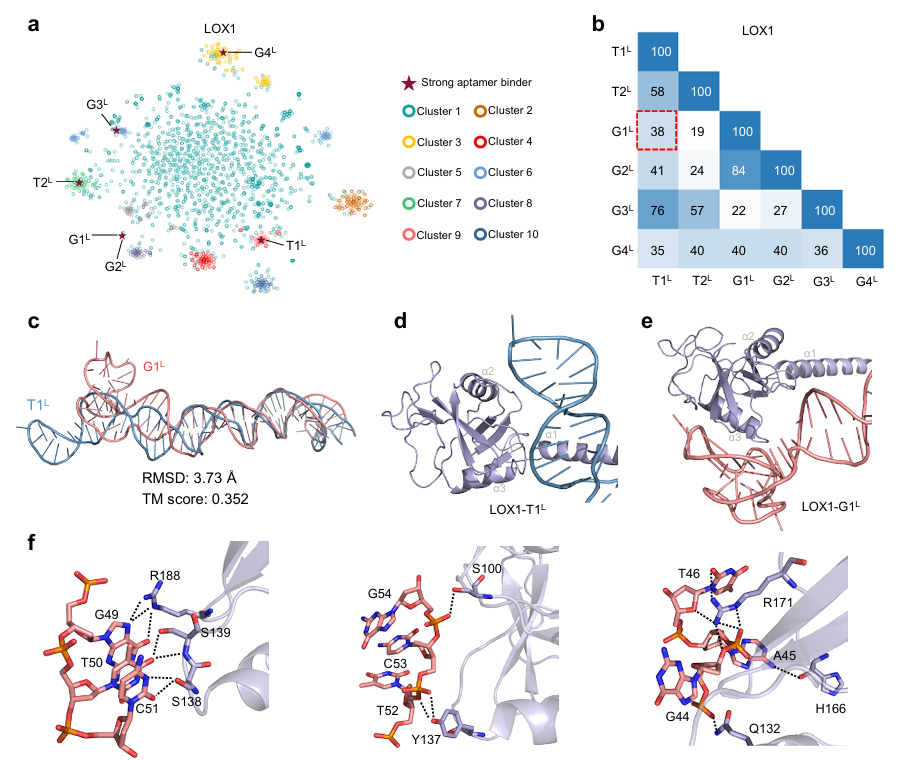
图三 生成适配体的序列多样性与三维结构特征
鲁棒性强:面对 latent 空间扰动,生成序列的 k-mer 频率和 GC 含量仍与真实序列高度一致;
兼容性好:可集成 DNABERT、Evo1、Nucleotide Transformer 等多种核酸大语言模型,均保持优异性能;
泛化性广:成功设计蛋白酪氨酸激酶 7(PTK7)结合适配体,适配多种功能核酸设计场景。
四、功能核酸设计进入 AI 驱动时代
InstructNA 的出现,不仅解决了功能核酸设计中 “序列空间大、结构依赖强、效率低下” 的核心痛点,更建立了一套 “数据驱动 - 模型生成 - 实验验证” 的全新设计范式。其科学意义深远,首先在技术层面突破了传统方法的瓶颈,无需三维结构即可从头设计功能核酸,大幅降低了设计门槛,显著缩短了研发周期,让功能核酸的高效设计成为可能;其次在序列探索层面,生成的序列与传统筛选序列相似度低,成功探索了全新的功能核酸序列空间,为发现具有新颖结构和功能的分子提供了重要途径;最后在应用层面,该框架可广泛应用于适配体、核酶、基因调控元件等多种功能核酸的设计,将有力推动临床诊断、药物开发、生物传感等领域的技术革新,为相关领域的研究提供强大工具。未来,随着核酸大语言模型的持续发展和更多类型功能核酸数据的积累,InstructNA 有望进一步整合三维结构预测和分子动力学模拟,实现更高精度的功能核酸设计,让 “按需定制” 功能核酸成为常态。
五、总结
功能核酸的设计曾长期受制于序列空间庞大和结构 - 功能关系复杂的双重挑战,传统方法难以兼顾效率、活性和多样性。InstructNA 框架通过核酸大语言模型与高通量 SELEX 的创新融合,打破了这一僵局 —— 它以毫秒级速度生成高活性、高多样性的功能核酸,在转录因子结合 DNA 和蛋白适配体设计中展现出远超传统方法的性能,部分适配体的结合亲和力达到纳摩尔级别,且序列相似度低至 38%。这项研究不仅为功能核酸设计提供了高效、通用的全新工具,更标志着功能核酸设计正式进入 AI 驱动的新时代。随着技术的不断迭代和拓展,InstructNA 必将在生物医学、生物技术等多个领域发挥更大作用,推动更多功能核酸类产品的研发与应用,为人类健康和科技进步做出重要贡献。
论文链接:https://www.nature.com/articles/s43588-026-00965-3
| 





















