|

在蛋白质工程领域,设计具有特定功能的全新蛋白质序列是一项核心且极具挑战的任务。传统的生化方法耗时耗力,而深度生成模型的出现,为高效探索巨大的蛋白质序列空间带来了革命性希望。然而,现有主流模型,无论是自回归(AR)模型还是扩散模型,都普遍面临一个关键瓶颈:“分布中心化” 问题。它们倾向于学习并生成高概率区域的“主流”序列,却忽略了长尾分布中那些稀有但功能至关重要的蛋白质变体。
针对这一痛点,来自浙江大学的研究团队提出了一种开创性的解决方案——ProtFlow。这是首个将流匹配(Flow Matching, FM) 算法与大语言蛋白质模型(pLM)的语义空间深度融合的生成框架,旨在实现全面、高效的蛋白质语义分布学习与高质量序列生成。相关研究已发表在预印本平台bioRxiv上。
01 现有模型的困境:为何我们总在“主流”里打转?
蛋白质序列与自然语言的相似性,使得基于Transformer的自回归模型(如ProtGPT2)成为设计主流。然而,其“逐个预测下一个氨基酸”的生成模式,难以建模蛋白质中复杂的长程依赖和全局相互作用。更先进的扩散模型虽然能模拟全局相互作用,但其将噪声到数据的概率路径分割为多个独立小步,容易导致次优路径,使得生成样本依然聚集在训练数据的高概率区域。
这种“分布中心化”问题在功能蛋白(如抗菌肽AMP)设计中尤为致命。AMP的活性谱系通常呈现长尾分布,即对某些病原体(尤其是罕见或耐药菌种)具有高活性的肽段非常稀少。现有模型极易忽略这些稀有但关键的语义区域,导致生成的AMP多样性不足,广谱抗菌活性受限。
02 ProtFlow的核心创新:流匹配 + 蛋白质语义空间
ProtFlow的核心理念在于直接学习从标准高斯噪声到目标蛋白质数据分布之间的连续、全局最优概率路径。相较于扩散模型,流匹配能提供更直接的路径,理论上能以更少的采样步骤实现更全面的分布覆盖。
然而,直接将流匹配应用于离散的氨基酸符号空间会导致模型退化。ProtFlow巧妙地解决了这一难题:
1.语义空间嵌入: 利用强大的预训练蛋白质语言模型ESM-2,将离散的蛋白质序列映射到连续、富含生物学语义的隐空间中。这使得模型能够学习蛋白质的全局语义组织,而非仅仅关注局部的氨基酸组成统计特征。2.语义空间重构: 针对pLM嵌入存在的高维度和激活异常问题,ProtFlow设计了语义集成网络,通过压缩-解压缩模块对和归一化操作,重构出一个紧凑、平滑且稳定的语义隐空间,极大提升了训练效率和鲁棒性。3.整流流与Reflow技术: ProtFlow采用1-整流流(1-Rectified Flow) 算法,定义概率路径为直线,进一步简化并优化了流的学习。更引人注目的是,通过引入Reflow技术对模型进行微调,可以实现单步(One-step)高质量序列生成,将推理速度提升到极致。
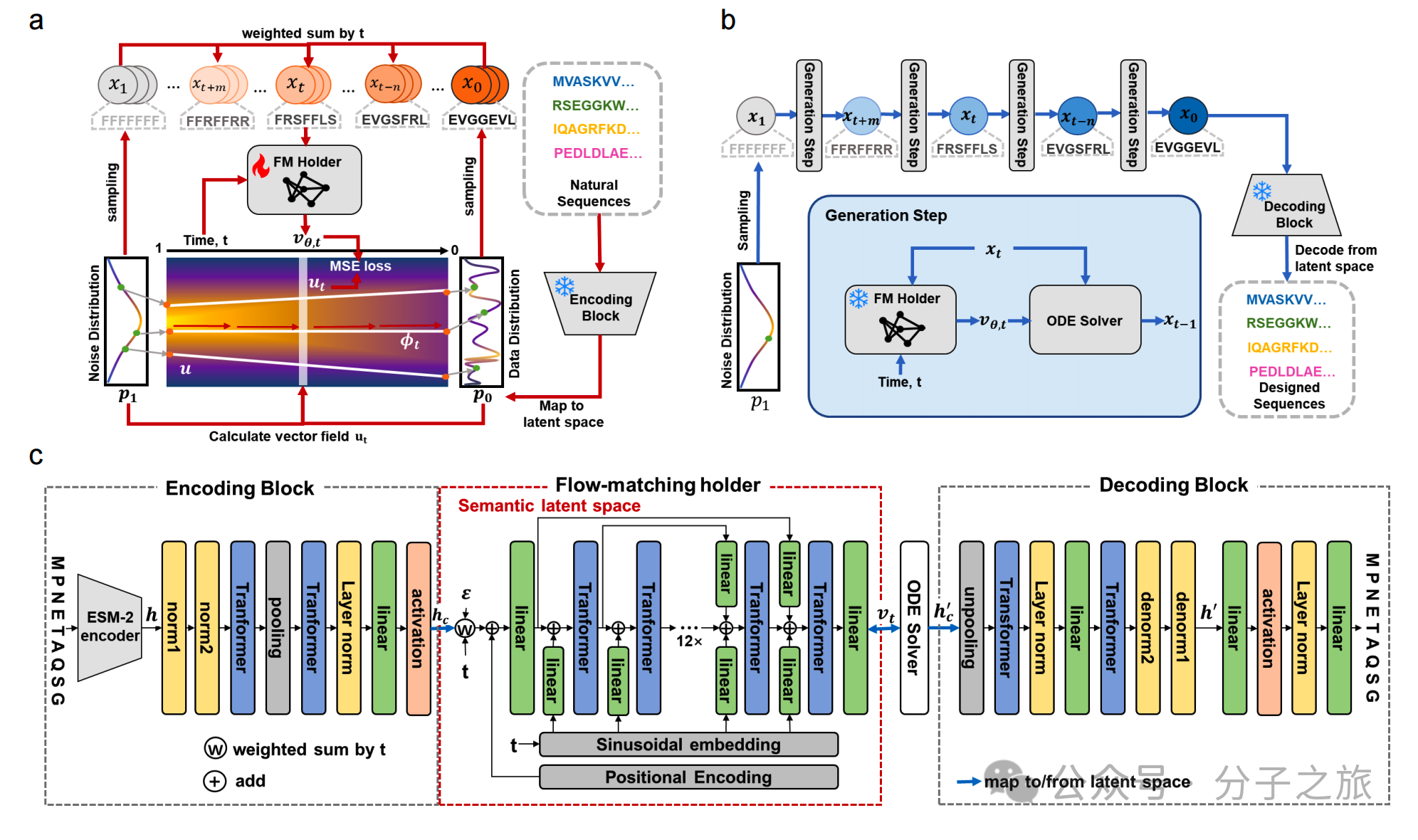
图 1:ProtFlow整体框架示意图,展示了从训练到推理,以及模型的核心架构
03 性能验证:全面覆盖,高效生成
研究团队在260万通用肽段数据集上预训练ProtFlow,随后在近1万条抗菌肽(AMP)数据上进行微调,并与当前最先进的多种生成模型(ProteinGAN, ProtGPT2, EvoDiff, DiMA, AMP-Diffusion, HydrAMP等)进行了全面对比。
1. 全面覆盖自然肽段分布
为了评估模型对训练数据分布的覆盖能力,研究人员使用ProtT5模型将生成的肽段映射到语义空间,并计算了弗雷歇距离(FPD)、最大平均差异(MMD)和最优传输距离(OT)。在所有指标上,ProtFlow均显著优于基线模型,表明其生成的序列在语义空间上与天然蛋白质最为接近。
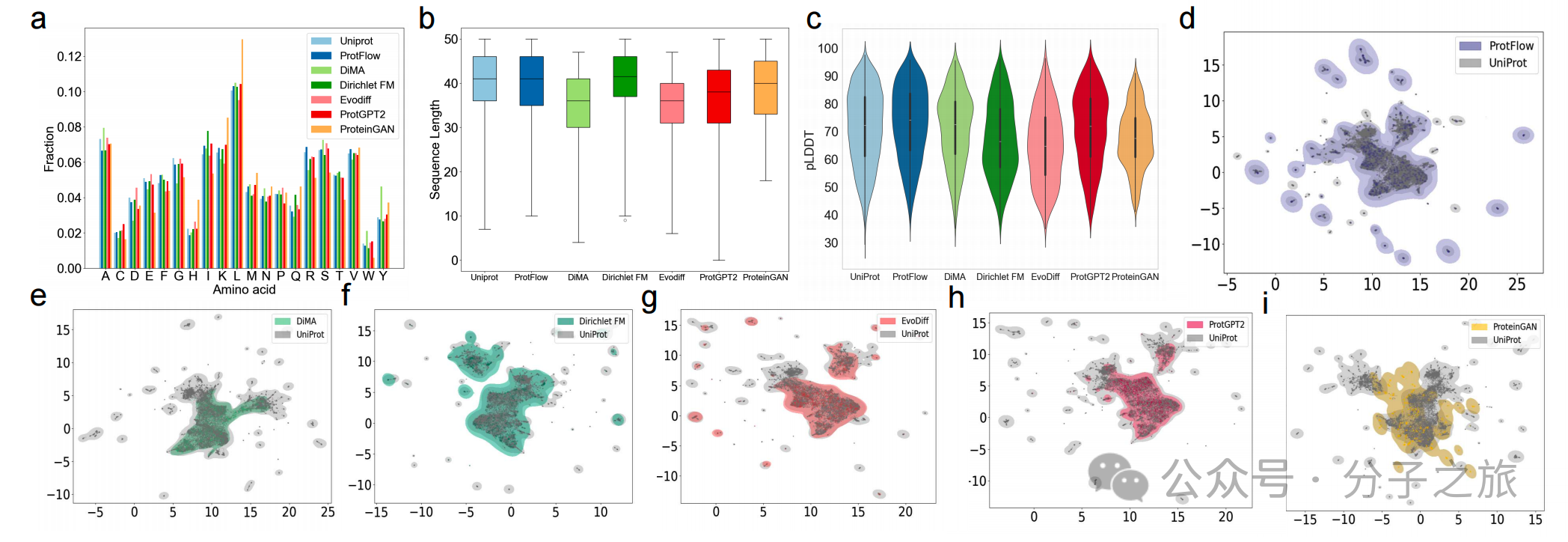
图 2:UMAP可视化显示,只有ProtFlow能够全面覆盖天然肽段
2. 生成高质量、可折叠的肽段
在序列层面,ProtFlow生成的氨基酸频率和长度分布与天然UniProt肽段高度一致。更重要的是,在结构层面的评估中,ProtFlow生成的肽段在pLDDT(预测局部距离差异测试)分数和TM-Score(结构相似性分数) 上均达到最高,同时保持了良好的结构新颖性(TM-Score未过高)。其ESM-2伪困惑度(ESM-2 pppl) 也最低,证明生成的序列最符合ESM-2从大规模数据中学到的自然蛋白质模式。
3. 设计高活性、广谱的抗菌肽(AMP)
在AMP设计任务中,ProtFlow(此时称为AMPFlow)展现出强大实力:
物化性质:生成的AMP在净电荷、疏水性、疏水矩等关键物化性质分布上与真实AMP高度吻合。
新颖性与多样性:在保证与真实AMP高度相似的同时,ProtFlow生成的肽段内部重复率最低,且与真实AMP库的重合度也较低,实现了相似性与新颖性的卓越平衡。
抗菌活性:使用三种主流AMP分类器(AMP-Scanner v2, Macrel, CAMPR4)进行评估,ProtFlow生成的肽段被预测为具有抗菌活性的比例最高,且其活性概率分布与真实AMP最为接近。
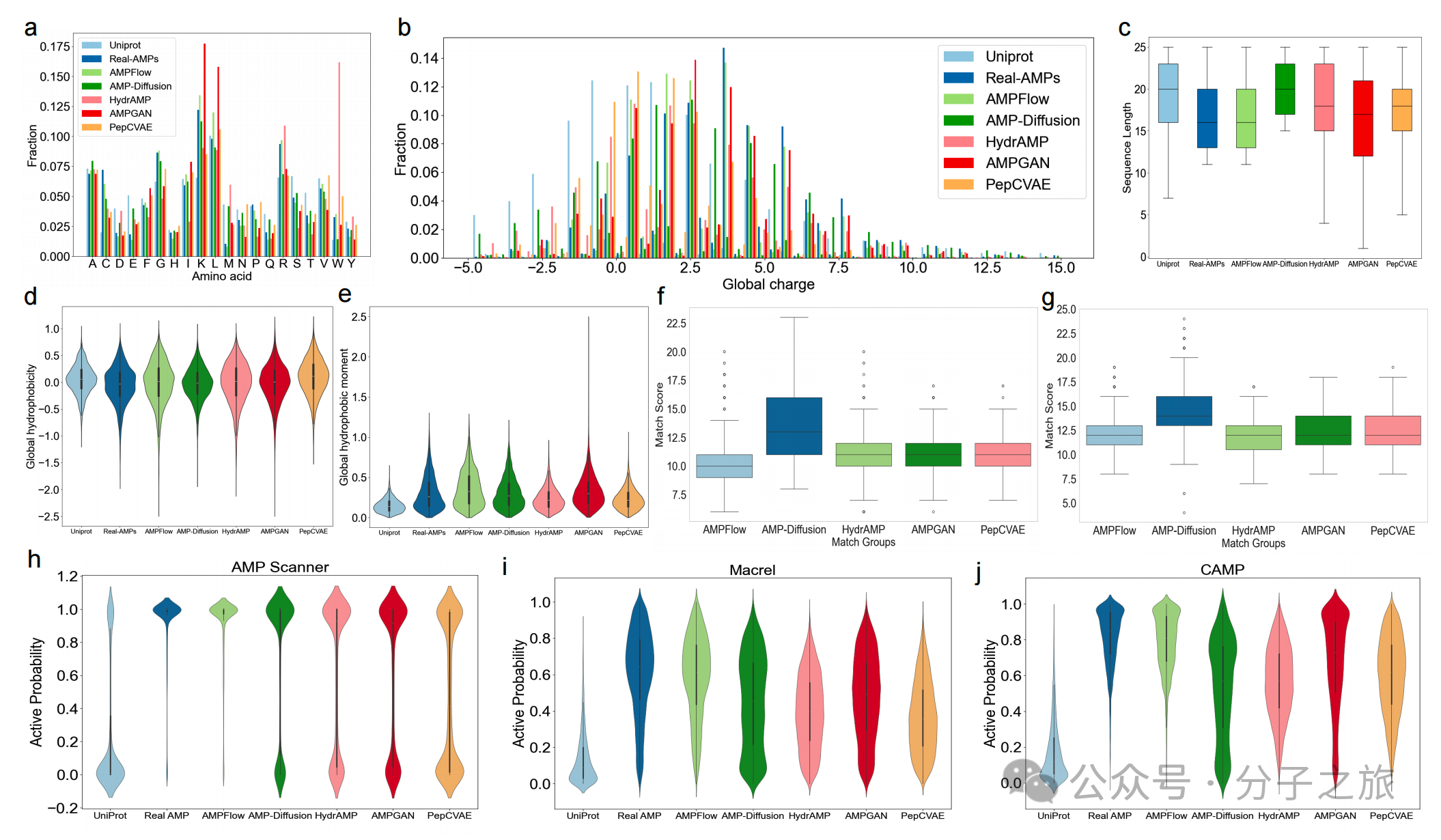
图 3:ProtFlow生成的抗菌肽(AMPFlow)在多种物化性质及活性预测分布上与真实AMP高度一致
4. 攻克“长尾分布”,有效覆盖稀有靶点
研究的关键验证在于:ProtFlow能否解决AMP设计中的“长尾分布”问题?为此,团队针对数据量最丰富的10种细菌训练了MIC(最小抑菌浓度)回归预测模型。结果显示,ProtFlow生成的肽段,对这10种细菌具有高活性(logMIC ≤ 1.5)的比例,最接近真实AMP的分布模式,平均达到了真实AMP活性比例的87.46%。尤其对于枯草芽孢杆菌(B. subtilis)、肺炎克雷伯菌(K. pneumoniae)和鲍曼不动杆菌(A. baumannii) 这些在基线模型生成中活性肽比例显著偏低的菌种,ProtFlow表现出了显著优势。这直接证明了其全面覆盖AMP语义分布、不遗漏稀有功能区域的能力。
04 总结与展望
ProtFlow的成功,标志着蛋白质序列生成模型从“拟合高概率区域”向“学习全面语义分布”的重要范式转变。其流匹配算法与蛋白质语义空间的深度融合,不仅解决了分布中心化难题,还实现了快速(甚至单步)的高质量生成。
该框架具有强大的通用性和扩展性。虽然本研究聚焦于抗菌肽,但ProtFlow可轻松扩展至信号肽、抗体、酶变体等多种功能蛋白的设计,特别是在数据有限或分布不均的场景下潜力巨大。未来,研究团队计划将其扩展至更长的蛋白质序列、多结构域蛋白,并探索与结构信息等多模态数据的结合,向着通用型蛋白质设计基础模型的目标迈进。
【文章原文】https://www.biorxiv.org/content/10.64898/2026.02.14.705870v1.abstract
文章改编转载自微信公众号:分子之旅
原文链接:https://mp.weixin.qq.com/s/1ATKD4lbbezhMJKfEa_qnA?mpshare=1&scene=1&srcid=03121GCYmctCyGq3xPCONqEg&sharer_shareinfo=3d877499cc9f70ce3557897b432b7506&sharer_shareinfo_first=3d877499cc9f70ce3557897b432b7506&from=industrynews&color_scheme=light#wechat_redirect | 





















