Janus-QUBO:一种兼具对偶性感知的基于可调量子启发式景观探索化学空间的框架
一、背景介绍
在广阔的化学空间中发现新分子是一项核心科学挑战,利用深度生成模型(DGMs)进行从头分子设计,已成为加速新型药物和材料发现的强大范式。通过在海量化学数据库上训练,利用变分自编码器(VAE)、Transformer等模型引入自参考嵌入字符串(SELFIES)等稳健的表示方法探索庞大的化学空间,生成具有期望性质的分子。目前的主流方法是将分子编码至高维连续潜空间(如高斯先验),并通过平滑的梯度指导性质优化。然而,基于连续潜空间构建的“黑箱”范式,在平滑优化景观与固有离散分子结构之间的根本性不匹配问题,往往限制了全局探索的效果。
为了克服该局限,本文提出了Janus框架,将分子设计重构为一个透明的、受物理启发的组合优化问题,不仅采用离散表示,而且能够以科学透明的方式释放其全局优化潜力。Janus的核心是一个带有正则化二进制瓶颈的基于Transformer的自编码器,可将分子映射成紧凑的离散潜空间,这种表示方式使得分子生成与优化问题能够被重构为二次无约束二进制优化(QUBO)问题,进而解锁多种协同功能。
在分子生成方面,Janus利用经典和量子退火器高效遍历结构化能量景观,实现化学骨架的全局发现。关键的是,在分子优化方面,该框架通过利用可量化的特征交互作为机器学习发现的SAR规则,这摆脱了盲搜模式,能够通过选择性修改潜空间比特位实现合理、可解释的优化。与当前最先进方法的基准测试结果表明,该方法在保持骨架完整性的同时实现了更优的多目标性能,避免了启发式基线方法中常见的结构碎片化问题。
同时该论文在专用量子计算机上验证了工作流的可行性,并证明了其在类药性质优化中的有效性。Janus将强大的组合探索与深度模型的可解释性相结合,建立了一个合理的、稳健的量子辅助分子设计框架。
二、技术框架
本文提出的Janus框架,将生成建模与组合优化统一为一个协调的、兼具对偶性感知的系统。从结构上看,Janus以稳健的数据基础为起点,通过使用比例采样策略构建了一个大规模、化学多样性的数据集(图 1a);随后基于Transformer编码器将这些分子映射为潜表示(图 1b);关键的是,本论文使用Gumbel Softmax离散化方法将该表示压缩为正则化的二进制编码(图 1c),这种表示过滤了噪声,避免了连续先验的拓扑不匹配问题,同时配对的解码器保证了分子的忠实重构(图 1d)。在功能上,Janus利用因子分解机(FM)构建了QUBO能量面,充当 “导航器”(图 1e),通过显式建模二进制潜特征之间的成对相互作用,该模型将分子发现从盲目的随机搜索转化为透明的、由物理驱动的能量最小化过程。
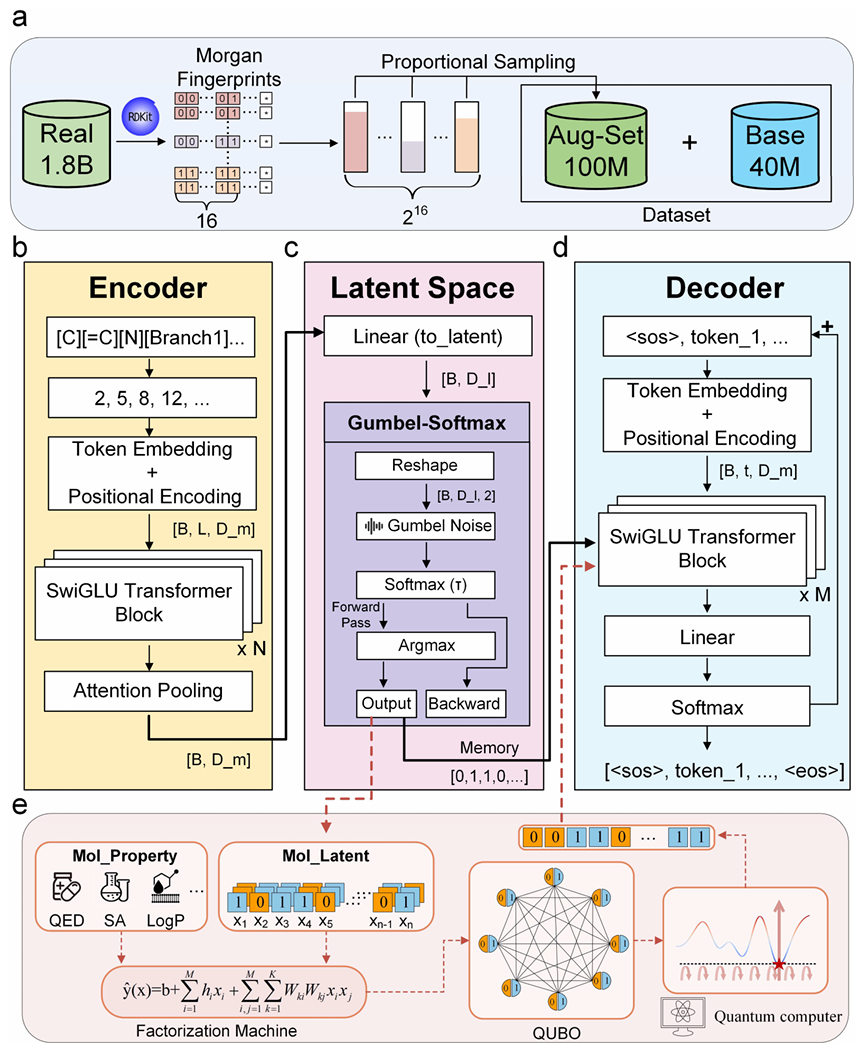
图1 分子生成重构为QUBO问题的Janus框架概述
a.基于Morgan指纹的比例采样方法,从18亿个已知化合物中构建大规模、多样性的数据集;
b.编码器模块,使用Transformer架构将分子的SELFIES字符串映射为连续潜表示;
c.潜空间模块,通过Gumbel-Softmax技术将连续表示离散化为固定长度的二进制编码;
d.解码器模块,从离散二进制编码中重构分子的SELFIES字符串;
e.优化模块,因子分解机学习二进制编码与分子性质之间的关系,将设计任务构建为QUBO问题。
2 结果
2.1 分子从头生成中性质的基准评估
Janus框架在生成具有理想类药性质的分子方面显著优于已有的基准方法(表1),其优越性在药物相似性定量评估(QED)、合成可及性(SA)、辛醇-水分配系数(LogP)、Lipinski五规则(Ro5)等所有关键指标上均保持一致。无论是在全部生成的分子群体中,更关键的是,在前5%、10%、20%候选分子中均表现出高性能,这一结果凸显了该框架识别高质量分子的有效性。
在Janus框架的不同变体中,Janus-FM和Janus-RBM展现出独特且互补的优势:Janus-FM生成了具有最高药物相似性的分子,在所有性能层级上均实现了优越的QED评分,且生成的化合物中处于理想logP范围([1,5])的比例显著更高;而Janus-RBM则在合成可行性上表现突出,产生了最低的平均SA分数。与之相对,Janus-BER变体(无指导随机采样)作为对照基线,其性能显著偏低,这证实了尽管二进制潜空间经过正则化处理但仍具有复杂性,需要通过能量景观(FM或RBM)进行定向导航,才能挖掘其中的高质量区域。上述两种模型均实现了近乎100%的Ro5规则符合率,证实其与药物化学的基本原理相契合。
与之形成鲜明对比的是,MolGPT、JTVAE等基准模型生成的分子库性质表现较差,不仅在识别高分候选分子的能力有限,在LogP、Ro5符合率等约束性指标上也表现不佳。总体而言,这些结果证实了Janus框架的优越性。
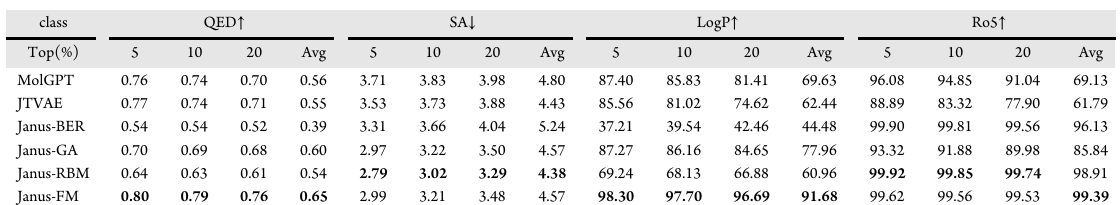
说明:
(1)对生成分子中按性质评分排名的前5%、10%、20%进行评估,“Avg”列为全部生成分子的平均评分;
(2)↓表示指标值越低越好,↑表示指标值越高越好,每个指标的最优性能以粗体显示,LogP分数表示具有位于所需范围[1,5]内值分子的百分比,Ro5分数是满足特定结构规则的分子百分比;
(3)Janus-BER、Janus-RBM、Janus-FM分别是以伯努利分布、受限玻尔兹曼机、因子分解机作为代替属性预测器的模型变体。
2.2 高质量化学空间的探索与覆盖度
通过分析千万量级生成分子在QED和SA Score上的概况,本论文评估了各模型对高质量化学空间的探索能力。针对每个模型,该论文确定了其经验帕累托前沿,该前沿定义了模型达到的最优属性权衡。随后,论文采用两个指标对这些前沿进行评估:超体积(Hypervolume),即可访问的高质量属性空间的体积;覆盖度(Coverage),即高性能解决方案的多样性。
图 2a 总结了该多目标分析结果,也展示了各模型的不同能力:FM模型实现了最大的超体积和最高的覆盖度,表明其既覆盖了广泛的高质量化合物区域,还达到了性能权衡的极限;相比之下,受限玻尔兹曼机(RBM)模型的超体积更小,其分子分布集中在高QED和低SA评分的理想区域(图2a),这表明了其内在的生成偏好:RBM模型更倾向于生成性质均衡的类药化合物,而非具有极端性能特征的分子。
总之,帕累托分析强调了各模型的互补优势:FM模型擅长探索化学空间中的“稀有优质区域”,能够发现性能前沿的新型结构;而RBM模型则专注于生成一致性高、质量优的分子库。这种差异体现了Janus框架的适应性——通过选择合适的代理模型,生成过程可以针对不同的药物发现场景进行定制,适用于探索性先导化合物发现(FM)或靶向型化合物库设计(RBM)。
为了验证该框架的端到端可行性,该论文在专用量子计算机上运行了Janus-FM的生成过程,成功生成了一系列有效且新颖的分子。图2b展示了量子硬件生成的代表性化合物,证实了整个工作流的实际可行性。
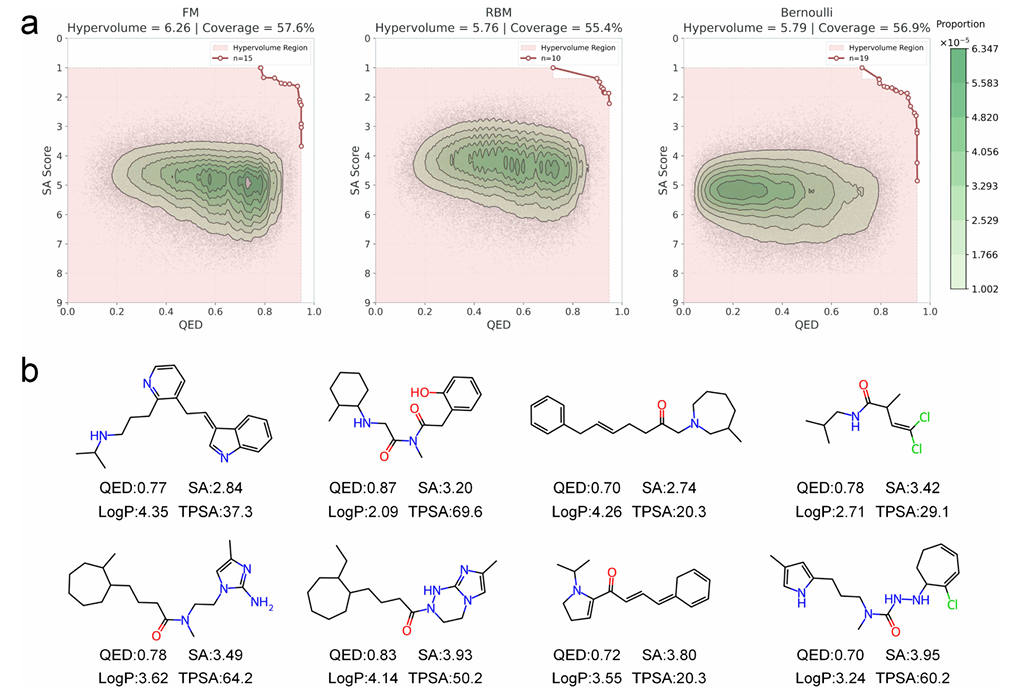
图 2 多目标性能分析及生成分子示例
a. 因子分解机(FM)、受限玻尔兹曼机(RBM)、伯努利模型(Bernoulli)三种模型的多目标性能分析,生成分子的分布在QED和SA评分空间中,红线为经验帕累托前沿;同时给出了各模型的超体积和覆盖度,用于量化最优解的质量和多样性;
b. Janus-FM框架通过专用量子计算机生成的代表性分子,验证了工作流的端到端可行性,并展示了各化合物的关键计算属性。
2.3 离散潜空间的可解释性与属性建模
Janus框架的一大关键优势是其高度结构化、可解释的离散潜空间。为了探究该空间与分子属性之间的关系,本论文开发了一个代理模型,可直接从128维二进制潜编码预测目标属性(图3a)。通过对该模型进行全局可解释性分析,将128个潜变量划分为四种不同的功能类型:部分变量被称为“增强器”,对属性始终产生正向影响;其他变量称为“抑制器”,对结果始终产生负向贡献。最值得注意的是,第三类变量充当“反转开关”,其贡献会随状态变化从正向转为负向(或反之,图3b)。在分析中,每个变量的影响大小与其到原点的距离成正比。
聚焦于影响最显著的变量,可以发现单比特位翻转可精确调控分子性质(图3c)。例如,激活一个“增强器”比特位会显著提高性质值,而翻转一个“抑制器”比特位则会导致性质值显著下降。该分析为高效的单比特位优化提供了清晰的策略。关键的是,本论文还发现多个潜变量之间存在强烈的协同效应:特定变量在共同激活时,产生的联合正向贡献远超过各自单独作用的累加效应(图 3d),这一发现揭示了潜空间中存在非线性的高阶相互作用逻辑。
最后,论文将这些高影响的交互特征整合至代理模型中,大幅提升了模型的预测性能(图 3e),体现在R-平方(
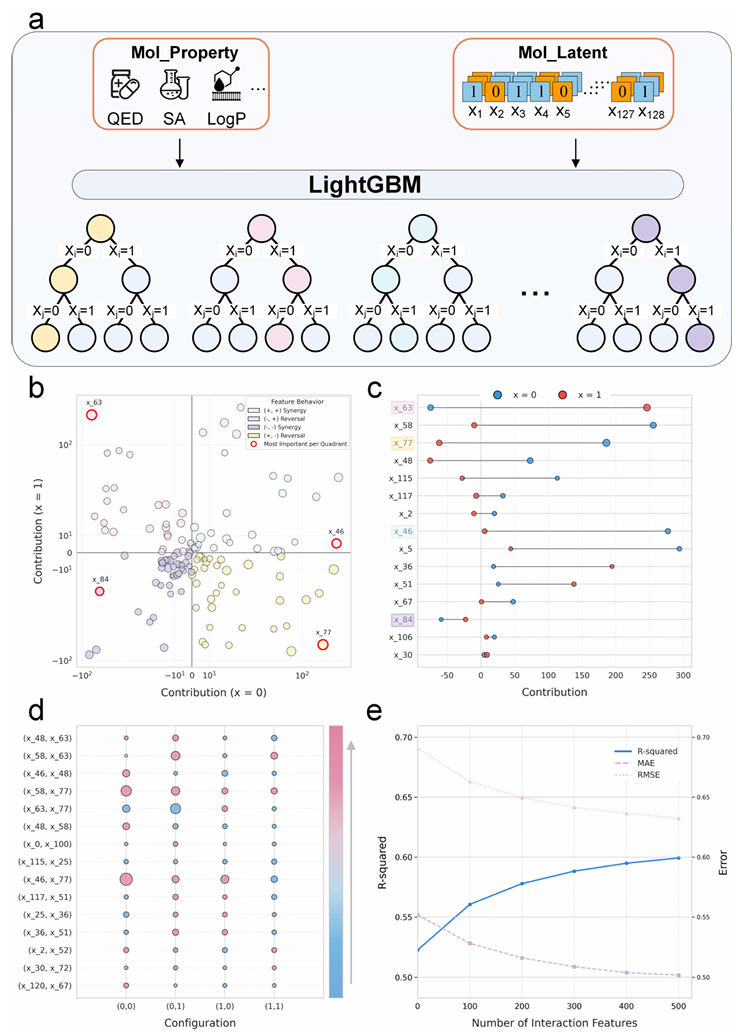
图 3 离散潜空间的可解释性与性质建模
a. 基于 LightGBM 的代理模型示意图,该模型可直接从 128 维二进制潜编码预测分子性质;
b. 散点图根据 128 个潜变量在关断(
c. 哑铃图展示了影响最显著的变量其比特位从 0(蓝色)翻转至 1(红色)时的性质贡献变化,量化了单比特位编辑的影响;
d. 对潜变量顶级配对之间的非线性相互作用效应进行可视化,展示了其在四种可能的二进制配置(
e. 代理性质模型的性能(以
2.4 化学逻辑的定量验证与优化基准测试
定量特征归因分析表明,潜空间具有连贯且符合化学规律的组织方式(图 4a,补充表 S2):与评分提升相关的潜特征(如
综合来看,这些发现表明潜特征的激活对应于结构化、与性质一致的亚结构调控,而非随机的相关性,凸显了模型在其潜空间中编码了符合化学意义的组织方式。
基准测试结果凸显了 Janus 框架在优化与结构保留之间实现平衡的卓越能力(图 4b、c,补充表 S3):在结构层面,Janus 在分子量(MW)、拓扑极性表面积(TPSA)、氢键受体数(HBA)等关键描述符上与参考分子高度契合,在雷达图的上半球形成了近乎完整的多边形;而 REINVENT4、GBGA等基线方法则表现出明显的特征收缩,表明其结构复杂性降低。分子量分布进一步印证了这一趋势(图 4c):Janus 的分子量分布与参考分子保持高度一致,而基线方法的分布则向低分子量偏移。在性质层面,Janus 在 QED 和 log P 指标上实现了显著提升,性能达到或超越当前最先进的方法。
综合来看,这些结果表明 Janus 框架避免了由碎片化驱动的无意义优化,而是实现了建设性优化,在提升类药性质的同时,保留了药物化学中关键的骨架特征。
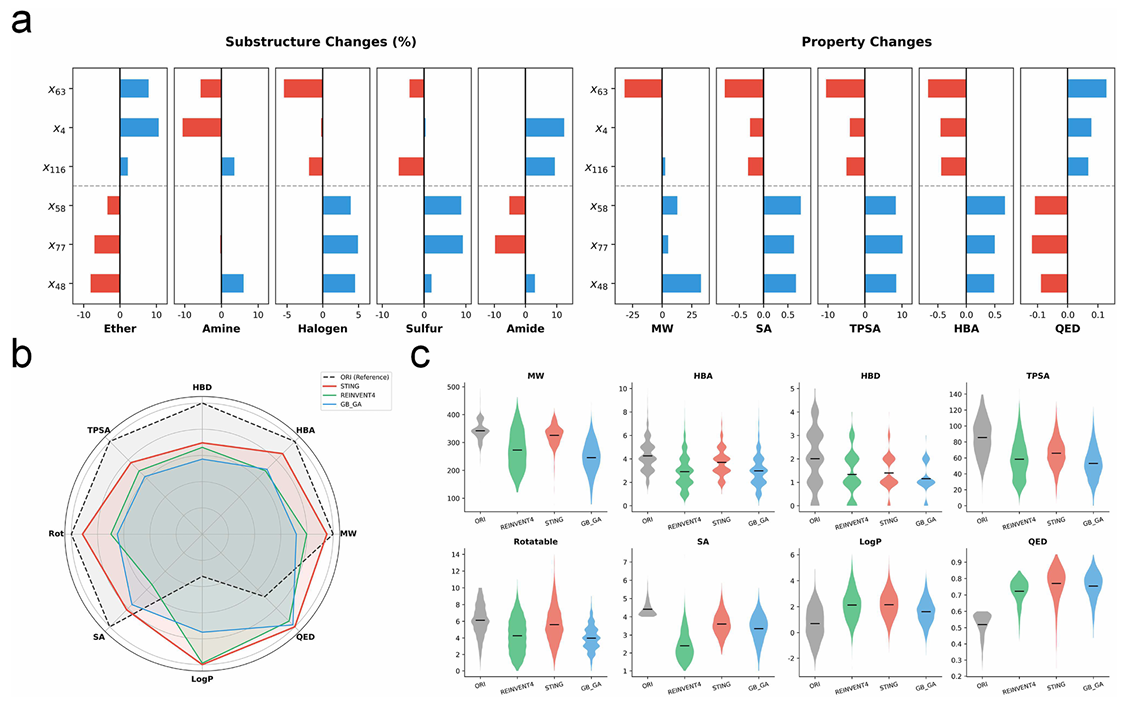
图 4 化学逻辑的定量验证与多目标优化性能
a. 特征归因图,展示了激活特定潜比特位引发的亚结构频率(左图)和物理性质(右图)的净变化;蓝色向右延伸的柱形表示正向变化,红色向左延伸的柱形表示负向变化;
b. 雷达图对比了 Janus 框架(红色多边形)生成的优化分子与原始参考分子(黑色虚线)的归一化结构和性质轮廓,基线方法(绿色 / 蓝色)的轮廓则出现收缩;纵轴和横轴分别归一化至所有方法中的最大值,Janus 框架在上半球体现出骨架保留性,在下半球体现出性质优化效果;
c. 小提琴图展示了八种分子描述符的密度分布。
2.5 案例研究:面向应用的分子优化
为验证框架在实际场景中的实用价值,我们开展了一项性质驱动的分子优化案例研究,将靶向干扰素基因刺激蛋白(STING)的初始分子转化为类药性质更优的优化变体。优化过程通过对分子的离散潜表示进行系统性修改实现,我们从两个尺度评估了该过程的有效性:宏观尺度,分析生成候选集中的性质分布;微观尺度,通过分子对接探究其结构变化和蛋白 - 配体相互作用。
宏观性质变化:结果证实,该框架可在保持结构多样性的同时,系统性提升多项类药性质。在候选集层面,优化后的分子在 QED、SA 评分、log P、TPSA 等关键指标上实现了协同提升(图 5a)。值得注意的是,QED 分布从低分区的集中状态转变为高分区的广泛分布,这一转变表明框架探索了多样的高质量骨架空间,而非收敛于单一模式。同时,SA 评分向高合成可及性方向偏移,说明我们的策略在提升其他性质的同时,拓展了可合成的化学空间。
微观结构分析:为了在生物学背景下说明这些能力,我们将两个具有代表性的分子与 STING 蛋白进行了对接。该分析表明,该框架能够识别并消除不利的结构基序,例如受张力限制的四元环,同时优化相互作用模式。例如,其中一个优化分子表现出较低的合成复杂度,并与靶标形成了更致密的氢键网络(图5b)。第二个案例展示了对氢键几何构型的优化;尽管氢键数量保持不变,但一个关键的相互作用距离缩短至更理想的范围,从而增强了结合特异性(图5c)。总而言之,我们的框架在化学可行性与生物功能两方面协同优化分子,为加速先导化合物优化提供了一条高效路径。
综上,我们的框架可协同优化分子的化学可行性和生物学功能,为加速先导化合物优化提供了高效途径。
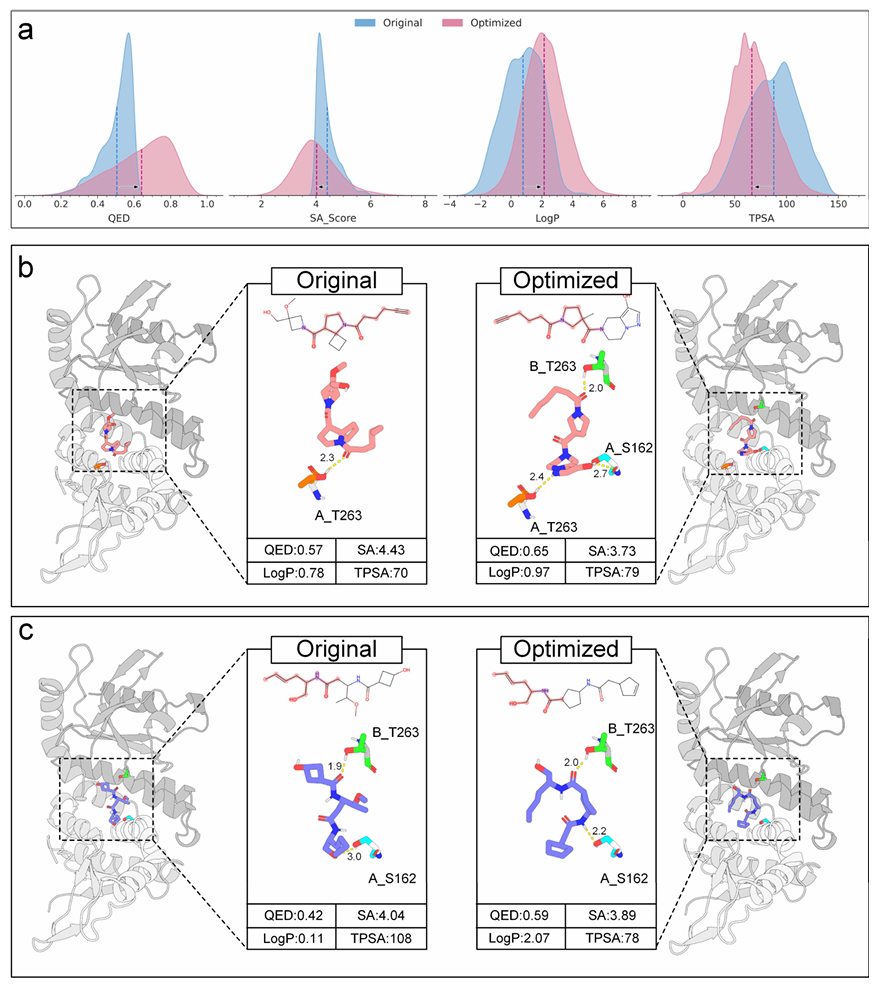
图 5 靶向 STING 蛋白的性质驱动分子优化案例研究
a. 初始分子库(蓝色)与优化分子库(红色)的关键性质(QED、SA 评分、LogP、TPSA)分布对比,所有指标均实现系统性提升,虚线为各分布的均值;
b. 初始分子与其优化变体的结合构象对比,优化后的配体消除了张力四元环,并与 A_S162 残基形成了新的有利氢键,形成了更致密的相互作用网络;
c. 另一分子优化实例,优化后的配体将与 A_S162 残基之间的氢键距离从 3.0 Å 大幅缩短至 2.2 Å,优化了结合模式,提升了相互作用特异性。
3 讨论与结论
Janus 框架首次将性质指导的分子优化重构为一个透明、可控且高效的闭环系统。我们的核心目标是通过系统化的工程框架,证明基于 Transformer 的自编码器与离散二进制潜空间相结合的协同效应,这一组合为将性质预测重构为 QUBO 问题提供了独特的可能。我们通过模型在生成高类药性质分子上的优异性能,以及在探索高质量化学空间上的稳健表现,验证了该方法的有效性。该框架的一大核心特征是其双重能力:既利用量子计算实现分子生成的全局优化,又能提供人类可解读的构效关系(SAR)规则,支持合理、可解释的分子设计。
离散二进制潜空间是本框架的基石,这一设计选择并非单纯为适配量子求解器,而是为克服连续优化 “黑箱” 本质的刻意策略。在连续空间中,性质优化依赖于通过神经网络代理模型进行梯度反向传播,而该代理模型是一个高度非线性的不透明函数,其景观无法实现全局可视化。通过将连续空间映射至因子分解机,我们将优化景观转化为 QUBO 模型,有效将神经网络的 “黑箱” 转化为 “白箱” 数学对象 ——
这一构建方式提供了一种全新的优化范式:不再是在平坦流形中沿梯度下降,而是利用基于物理的求解器遍历结构化的能量景观,这些求解器通过隧穿机制跨越能垒,探索化学空间的多样区域。我们在量子计算机上完成的概念验证,将这一设想落地实践:尽管量子生成分子的整体性质评分与经典模拟退火(SA)相当,但这一结果验证了从深度生成模型到 QUBO 模型的流水线的端到端可行性。关键的是,该实验证明量子硬件能够成功从我们生成模型定义的复杂分布中进行采样。
4 方法与数据
4.1 数据集制备
我们构建了一个大规模训练语料库,包含来自 Helixdock 的 4000 万个分子核心数据集,以及从 Enamine REAL 数据库中采样得到的 1 亿个分子扩充数据集。为保证化学多样性,我们实现了基于 Morgan 指纹的结构比例采样策略,平衡常见骨架和稀有结构基序的覆盖度(采样细节见补充方法 1)。所有分子均以 SELFIES 字符串形式表示。
4.2 离散潜空间自编码器(DLS-AE)
我们开发了基于 Transformer 的自编码器,将 SELFIES 字符串映射至结构化的二进制潜空间。
4.2.1 编码器
编码器采用由
4.2.2 潜空间离散化
为得到紧凑的二进制表示,我们采用 Gumbel-Softmax 方法,该模块将上下文向量投影为对数几率,并按公式 1 进行可微采样:
随后,通过公式 2 的操作得到最终的二进制潜编码
其中,
4.2.3 解码器
解码器与编码器结构相匹配,通过交叉注意力机制,以自回归方式重构分子。
4.2.4 训练目标
模型通过最小化复合损失进行训练:重构损失(
总损失为重构项和正则项的加权和,我们采用退火策略逐步提高 KL 权重
通过优化公式 3,模型在实现高保真度重构的同时,生成适用于下游任务的正则化离散潜空间(详细的架构和损失构建方式见补充方法 2)。
4.3 离散潜空间的性质建模
要在离散潜空间中实现高效的性质指导优化,需要一个将潜编码
4.4 面向量子启发式优化的 QUBO 构建
因子分解机的二次结构可自然映射为二次无约束二进制优化(QUBO)问题,目标函数如公式 4 所示,以哈密顿量形式定义:
其中,
4.5 向伊辛模型的转换
所得的 QUBO 问题可以通过多种经典或量子算法求解。为了与基于物理模型(如 simulated annealing)的求解器兼容,我们将 QUBO 问题转换为一个等价的 Ising 模型。Ising 模型使用自旋变量
耦合矩阵(for
外磁场:
利用由公式 5 和公式 6 得到的参数,我们使用 Kaiwu SDK (v1.2) 求解了所得的 Ising 模型。
4.6 可解释性分析
为解读生成器编码的化学设计规则,我们采用高保真度的梯度提升决策树(GBDT)模型(LightGBM)作为代理模型,建立离散潜空间与分子性质之间的映射关系。
4.6.1 代理模型训练
基于 LightGBM 框架训练 GBDT 回归模型,以 128 维二进制潜编码为输入特征,以分子性质评分为预测目标。
4.6.2 树结构预处理
首先,解析训练后的 LightGBM 模型,将其结构转换为数据框,数据框的每一行代表一个树节点,包含节点索引、父节点索引、分裂特征、阈值、子节点、叶节点值(预测值)和样本数。为提高遍历效率,我们还构建了哈希表用于快速查找节点。
4.6.3 模型解释的层级化框架
我们的分析框架通过三个层级解构模型的决策过程,每个层级采用特定的定量指标。
层级 1:量化单个变量的贡献
首先,我们量化了每个潜变量的独立贡献。我们使用公式 7 计算某一特征状态(
其中,
层级 2:衡量成对相互作用的重要性
接下来,我们识别哪些潜变量通过相互作用影响分子性质。我们为每一对
公式 8 的各组成部分定义如下:
- 基础评分
优先考虑高影响力的叶节点(较大的 )以及具有广泛样本覆盖的叶节点(较大的 )。 是该交互变量对中较浅节点的深度,衰减因子 (设定为 0.05)对更接近根节点发生的交互给予更高权重。 是该特征的总增益(来自 LightGBM),用于奖励在个体上具有较高重要性的特征之间的交互。
层级 3:解构特定的相互作用效应
最后,针对相互作用重要性高的变量对,分析其四种可能取值配置的效应:
公式 9 揭示了协同、拮抗等复杂的非线性关系,例如,配置(
这一集成的层级化框架将 “黑箱” GBDT 模型转化为包含可解释设计规则的 “白箱” 知识库,为性质驱动的分子优化奠定了稳健的基础。
4.7 面向分子优化的可解释性指导
利用这些可解释性见解,我们设计了基于知识的优化策略,与盲搜不同,该策略通过以下方式启发式地遍历景观:
单特征指导的优化
该策略假设,翻转一个影响显著的潜变量可实现性质的显著提升。我们识别出贡献评分最高的前 K 个潜变量(分析见 4.6.3 层级 1),对于给定的潜编码
本策略的目标是通过最小的结构修改(汉明距离为1)实现性质的大幅提升。
配对配置优化
该策略通过直接应用高影响特征对的最优配置来处理潜变量之间复杂的协同效应。我们基于 Section 4.6.3 Level 3 的分析,识别出前 K 个交互变量对
贪心累积优化
为探索更广阔的化学空间区域,我们采用贪心迭代优化策略:从初始解出发,在每次迭代中评估所有可能的单特征翻转和配对配置修改,随后应用使 QUBO 能量降幅最大的修改方式,将新的解作为下一次迭代的起点。我们执行两条独立的贪心搜索轨迹:一条基于单特征修改,另一条基于配对配置修改,框架将两条轨迹的所有中间解收集为候选变体。该方法构建了一条指向低能量区域的定向、逐步路径,有效追踪至局部最优的轨迹。
通过并行执行上述三种策略,我们的框架为每个初始分子生成了多样的候选变体集合(数十个),这些变体与原始分子的汉明距离各异,且具有多样的化学结构。最后,我们对所有变体进行去重,并按 QUBO 能量排序,得到高质量的候选分子列表,用于后续验证。并行计算加速了整个工作流,实现了高效的大规模分子优化。
数据可用性声明
本研究使用的 Enamine REAL 数据库可从公共渠道获取:https://enamine.net/compoundcollections/real-compounds/real-database;
核心数据集 HelixDock 可通过其官方网站申请获取:https://paddlehelix.baidu.com/app/drug/helix-dock/forecast;
本研究开发的源代码在 GitHub 开源:https://github.com/AdingWonderfulAdventure/Janus-QUBO;
训练后的模型及相关数据已上传至 Zenodo:https://zenodo.org/records/17647169;
本论文链接:点击访问文章