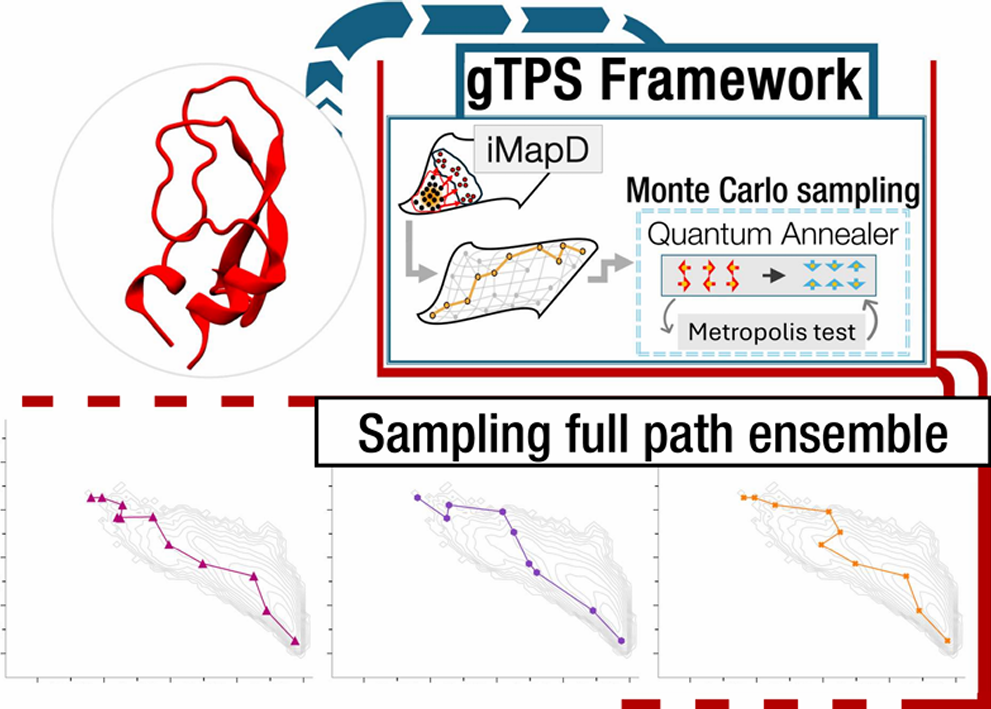
大分子不同构象转移路径预测
本研究主要基于图路径采样(gTPS)方法,结合分子动力学模拟和量子退火技术,从代表构象聚类和图构建出发,估算关键中间态的动力学寿命与路径作用,精确采样蛋白质稀有构象跃迁路径。
一、背景介绍
1、大分子构象转变的生物学意义
生物大分子(如蛋白质、核酸等)在执行生命活动时,会经历复杂的结构重组过程,这些构象变化是其功能实现的基础机制。蛋白质通过折叠形成特定的三维结构来发挥生物功能,如酶催化、信号转导和分子识别,而这种折叠过程受到配体结合、聚合状态等多种因素的精密调控。
深入理解大分子构象转变的动态路径与分子机制,对于揭示生命活动本质、解析疾病发生机理(如蛋白质错误折叠导致的神经退行性疾病)以及指导创新药物设计具有不可替代的科学价值。例如在药物研发领域,许多靶点蛋白存在多种功能状态,小分子药物可通过选择性稳定特定构象状态来调控蛋白功能,这使得构象转变路径中的关键态成为药物设计的重要靶点。
2、经典分子动力学(MD)采样的局限性
分子动力学(MD)模拟可以深入了解大分子系统的结构和动力学特性。原则上,这些模拟可以阐明复杂的热激活构象转变所涉及的物理化学机制,并预测它们的动力学和热力学特性。
然而在实践中,它们通常受到采样问题的限制,即“采样瓶颈”——需要克服较大的自由能障碍以收集连接给定初始和最终构象状态的统计显著数量的过渡轨迹。MD模拟在亚稳态下的热波动上投入了指数级的大部分计算时间,但跨越能量障碍的事件稀少且难以捕获,使得对罕见跃迁事件的表征非常耗时。
3、过渡路径采样(TPS)的优势与挑战
过渡路径采样(Transition Path Sampling, TPS)技术专注于生成反应物与产物之间的完整过渡轨迹,无需引入非物理加速力或依赖特定的集体变量选择。其核心思想是在过渡态附近发起MD试验轨迹,并根据Metropolis标准接受或拒绝。
TPS的优势在于:
(1)只需定义初、末态,即可直接采样过渡路径;
(2)保持动力学的物理真实性,不引入人为偏置。
然而,TPS在实际应用中面临两大挑战:
(1)低接受率:试验轨迹的接受率高度依赖于MD初始条件的选择。对于复杂转换,接受率往往很低,极大地增加了采样成本。尽管有研究将TPS与机器学习集成以优化初始条件生成,比如在膜蛋白组装等复杂过程中展示了潜力,但仍需大量无偏仿真来获取过渡持续时间分布。
(2)强自相关:TPS产生的马尔可夫链在过渡通道空间可能出现强自相关。例如,在简单二维能量景观中,若反应物与产物之间存在两条能垒高度不同的通道,马尔可夫链可能在一个通道内徘徊较长时间,采样另一路径的切换频率极低,延缓了多通道采样的效率。
4、模拟退火采样与图路径采样(gTPS)框架
将分子动力学(MD)模拟与用于未知流形探索的无监督方案(如iMapD)和模拟退火(Simulated Annealing,SA)相结合,以克服采样瓶颈。在此方案中:
(1)流形探索与区域划分:首先在经典计算机上使用iMapD算法对分子构型空间的本征流形进行初步探索。在此过程中,每个生成的构象被视为一个有限区域,其空间尺度
(2)粗粒度网络模型:将这些有限区域作为无向图的节点,并基于简化的朗之万动力学(Langevin dynamics)理论推导出节点之间的转移概率表达式,从而计算连接初始区域与目标区域的一条给定序列的总体通量;
(3)QUBO建模:对构建的无向图进行QUBO建模,具体来说即是将每个节点和边赋予二进制变量;
(4)模拟退火采样:在构建的图上利用模拟退火手段并行生成过渡路径。具体而言,将温度调控视为图上能量权重的调节参数,通过随机动力学蒙特卡罗生成试验路径,并根据Metropolis准则进行接受/拒绝,以在不同区域间高效穿越能量障碍,减少路径自相关;
(5)统计校正:对采样得到的路径集合在经典计算机上应用Metropolis接受/拒绝步骤进行重加权,确保最终分布与底层朗之万动力学一致。
5、量子计算的实施方式
本研究已有量子退火技术的实现先例,鉴于相干光量子计算机在求解优化问题上的潜在优势,后续工作将探索将gTPS框架迁移至相干光量子计算机(以下简称CIM)平台,以进一步提升采样效率并扩大体系规模。
6、工作目标
本研究旨在通过将分子动力学(MD)模拟与无监督流形探索算法(iMapD)和量子计算相结合,构建图路径采样(gTPS)框架,以高效捕获和表征蛋白质稀有构象转变通道,并验证该方法在经典计算资源下的可行性与性能,为未来在相干光量子计算机(CIM)上进一步加速和扩展奠定基础。
本研究结合JCTC发表文献“Sampling a Rare Protein Transition Using Quantum Annealing”,以期实现大分子不同构象转移路径预测效果优化,并与D-wave方法进行对比。
二、现有方法调研
在大分子(如蛋白质、核酸)构象变化研究中,有效预测其转移路径依赖于两个关键环节:一是构象空间的充分采样,二是过渡路径的高效识别与建模。以下将分别综述目前主流的构象采样方法和过渡路径采样方法。
(一) 构象采样
构象采样旨在高效探索生物大分子的自由能景观,克服传统分子动力学模拟因时间尺度限制而无法跨越能垒的缺陷。
典型方法综述
| 方法 | 核心思想 | 优点 | 局限性 |
|---|---|---|---|
| 分子动力学(MD) | 牛顿方程模拟时间演化 | 原理清晰,物理可靠 | 跨越能垒能力弱,效率低 |
| 高温MD / 加速MD | 提升温度或平坦化势能面 | 增强跃迁能力 | 热涨落失真,影响路径真实性 |
| Replica Exchange MD (REMD) | 多温度体系并行采样并交换 | 扩展采样范围 | 系统大时开销巨大 |
| 元动力学(Metadynamics) | 在CV上加入历史偏置势能 | 推出系统跳出能垒 | 依赖CV选择,非通用 |
| 伞采样(Umbrella Sampling) | 强制系统驻留在指定区间 | 局部分布估计准确 | 需预先定义反应路径 |
| 玻尔兹曼发生器(Boltzmann Generator) | 用神经网络直接生成平衡构象 | 样本多样、快速采样 | 建模目标为状态分布,非动力学 |
| Diffusion Map / iMapD | 无监督学习构象流形结构 | 发现未知方向、无CV依赖 | 构建算法较复杂 |
(二) 过渡路径采样方法拓展
过渡路径采样关注状态A到状态B之间的构象跃迁机制,目标是生成具有物理意义的反应路径,并捕捉关键中间态,反映体系从初态到末态的典型演化方式。
2.1 主流方法综述
| 方法 | 原理 | 特点 | 局限性 |
|---|---|---|---|
| 过渡路径采样(Transition Path Sampling ,TPS) | 在路径空间上做Monte Carlo采样 | 物理性强,不需CV | 初始路径获取难,采样效率低 |
| 前向通量采样(Forward Flux Sampling ,FFS) | 将路径空间分段,从前向推进 | 可估速率,适合稀有事件 | 依赖界面设置,样本偏差大 |
| 弦方法 / 镇定弹性带(String Method / Nudged Elastic Band ,NEB) | 优化路径能量或CV变化 | 求最小作用路径 | 通常只得到单一通道,非统计采样 |
| 主导反应路径(Dominant Reaction Pathways ,DRP) | 基于最小action原理 | 高效估计路径 | 无法提供动力学信息 |
| 马尔可夫状态模型(Markov State Model ,MSM) | 将构象离散为状态网络 | 可还原动力学过程 | 依赖长时间轨迹,马尔科夫性假设强 |
| 里程碑法(Milestoning) | 将路径分解为多个小段 | 提高动力学信息统计 | 面临路径依赖问题 |
2.2 方法拓展与新兴技术
(1)机器学习增强TPS(Machine Learning-guided TPS):通过变分自编码器(VAE)等方式学习路径空间流形,提升TPS中“shooting move”的成功率与多样性;
(2)基于图的TPS(Graph-based TPS,gTPS):将构象与跃迁映射为图结构节点与边,将路径优化转化为图遍历问题(如Hamiltonian路径搜索),并可进一步映射为QUBO问题在量子退火器(D-wave)上求解,对路径空间并行搜索,跳出经典采样中的路径局部陷阱,在小系统或粗粒度表示下展现出优越的采样能力。
(三)总结
综上,现有构象采样方法与路径采样策略虽各具优势,但也面临显著的局限。例如,传统分子动力学及其变体在自由能面复杂的大分子体系中仍难以穿越高能垒;基于集体变量的增强采样方法对CV选取高度敏感;而路径采样方法如TPS与FFS则存在采样效率低、路径单一、初始依赖性强等问题,难以满足多通道路径识别与结构动力学相结合的需求。
相比于传统方法,本项目方法具备以下优势:
- 无需集体变量设定,天然适配复杂自由能面;
- 路径采样全局性更强,易于识别多通道与短程跃迁;
- 支持与量子类硬件无缝集成,具备可拓展性与前沿性;
- 在前期测试中(基于SA)已验证其物理合理性与路径可行性,为后续CIM引入奠定基础。
三、项目流程介绍
(一)未知配置空间探索
与其他流行的增强采样方法类似(尤其是Metadynamics),iMapD的效率来自于这样一个策略:尽量减少在已访问区域的模拟时间,把资源集中在新区域的探索上。
但iMapD无需选择collective variables(CV,集体变量),也不需要加入任何外部偏置力——无监督,而是使用DMAP(the Diffusion Map)“扩散映射”降维算法——以实现降维并推断内禀流形的局部结构。
接着,iMapD利用从Diffusion Map得到的几何信息,生成新的初始构象。从这些构象出发进行短时间MD模拟,就有很大机会探索到流形中动力学上可达的新区域。
iMapD算法的主要步骤如下:
步骤一:初始化构象集(短程MD)
从一个分子
步骤二:降维分析DMAP
DMAP所保留的维数应该与系统构象空间中“有统计意义的子流形维度”一致。为了简化方法,直接将构象投影到前两个“非平凡”的DMAP主分量上(也就是取二维降维结果),“非平凡”表示排除掉恒定项(如第一个为常数的特征向量)。
步骤三:选取边界点+构建边界邻域
从初始构象集中找边界点:
对每一个边界点,定义一个与其相邻的“邻域集合”
本项目在应用于蛋白动力学的实际设置中,将每个边界邻域的大小设置为30个点。每个边界邻域中,通过平均所有30个构象的笛卡尔坐标,得到一个代表性构象
步骤四:邻域内的PCA分析
对每一个边界邻域
将每个边界邻域中的构象投影到上述主成分构成的低维子空间中:
步骤五:Polar Star shooting的核心操作
利用先前在PCA子空间中构建的边界方向,从已知构象边界向外“延伸一步”,得到一个新构象点
新的、尚未探索的构象
其中,
步骤六:新点映射回原始空间(逆PCA)
将在PCA降维空间中构造的新点
步骤七:能量最小化
用
其偏置力的数学形式为:
其中:
对于反应坐标和接触图的计算如下:
其中:
对
在本项目中,我们使用JAX自动微分获得偏置力梯度代替真实力场。
步骤八:将新结构加入数据集,继续下一轮 iMapD
(二)本征流形上随机动力学的粗粒表示
1.图构建方法
如果对构象全集直接进行聚类,时间复杂度将很高。本项目采用FPS算法(Farthest Point Sampling,最远点采样)采出初始构象集中分布最广的自定义个数的点,在此设为200个。对选取出的构象,运用HC-UMPGA(Hierarchical Clustering with Unweighted Group Pair Method using Arithmetic Mean,一种层次聚类)从探索的全部构象集中找出代表性集合:
利用这
把每个代表构象
构建粗粒度无向图,首先只在配对
a. 节点度数 < 2 的从该点连接到其 RMSD 最近的两个邻居(即便 RMSD > d_cutoff);
b. 出现多个连通子图时且之间不连接时,与图中其他区域通过“最小RMSD的点对”建立连接,确保图的连通性。
2.路径概率理论推导
这种粗粒度理论是从连续体中的基础微观描述开始,通过计算每个代表性构型周围的扩散动力学的局部平均值得出的。路径积分形式用于连接这些局部信息并为反应轨迹分配统计权重。在下文中,我们将简要回顾这一理论。
从这样一个假设出发:在连续构象空间中的蛋白质动力学,可以用一个过阻尼Langevin方程来描述:
Langevin方程是统计物理和分子动力学中描述受热涨落影响的粒子运动的微分方程,它结合了确定性力和随机噪声项,用于模拟系统在热浴(如溶剂)中受到摩擦和随机碰撞的情况。
其中:
表示热噪声在不同时间之间是无关的(δ函数相关),系统的扩散能力与噪声强度是统一的;
原始的Feynman传播子定义为:
表示粒子从
其中,
被称为有效作用量,是在朗之万方程下定义的一个泛函,用于衡量某条随机路径的“代价”或“可能性”。
以上方程适用于连续体公式,因此不能直接用于估计粗粒度轨迹的相对权重。所以文献中通过诉诸重整化群论方法来计算底层微观朗之万动力学中的局部平均值,明确地推导出了粗粒度路径
在gTPS中,每条路径可以表示为整数序列:
其中
其中
其中:
然而,在实际应用中,对粗粒度路径概率采用不同的表达式可能很方便。注意到在同一区域保持时间间隔
引入一个统一假设:所有节点之间的平均跳跃时间一致。
那么从时间离散进一步转换为Hamilton–Jacobi (HJ) 形式的路径概率的空间离散化:
其中:
Hamilton-Jacobi 形式提供了一种估计过渡路径时间的简单方法。在这种形式中,系统沿着给定的粗粒度轨迹从区域 0 过渡到区域 M 所需的平均时间为:
引入近似值以获得更简单形式。为此,我们将所有距离
其中
3.图边权设置
所以在构建的粗粒度无向图中,边的权重如下定义:
基于时间离散:
基于空间离散:
权重对称是因为该方法只捕捉转移路径,不反映停留时间长短,也不反映路径之间的真正转移概率。如果需要恢复方向性,可用MSM方法来计算转移概率。
由之前的推导:逃逸率
(三)通过模拟退火对转移路径进行采样
QUBO转化:
每个节点
目标函数
其中:
分别为起点、终点和中间节点约束。
起点约束要求路径必须经过起点并且与起点有且仅有一条边连接。
终点约束要求路径必须经过终点并且与终点有且仅有一条边连接。
中间节点约束要求路径中除了起点和终点的每一个节点都必须只有两条边连接。
利用以上方式构建的QUBO模型,应用模拟退火算法进行大分子不同构象转移路径的采样。找出构象之间满足条件的可行路径。
在本项目中,可行路径的筛选阈值被设置为:满足所有约束的路径集中,路径总代价不超过最优路径代价的两倍的所有路径。
(四)路径重加权(暂未实现)
在使用广义 Ising 模型哈密顿量
为了纠正这种偏差,引用了一种基于 Metropolis 接受/拒绝准则的重新加权(reweighting)方法,使得最终采样的路径服从正确的统计分布。
这是 Metropolis-Hastings 算法中的接受概率,用于决定是否接受从当前状态
由于在对结果的分析中发现路径的质量都很接近,所以预计重加权也不会显著改变过渡路径系综分布。所以未实现该方法。
四、实验结果
(一)实验体系选择说明
本项目选取的蛋白质各项重要指标如下所示:
| 蛋白质 | 2pbi-Chain A | 1k5n-Chain A | 2hba-Chain A |
|---|---|---|---|
| 长度 | 424 aa | 276 aa | 52 aa |
| 最低 TM 分数介于起始和最终构象 | 0.447 | 0.630 | 0.683 |
| 最低 TM分数介于最发散构象 | 0.446 | 0.608 | 0.606 |
| 平均 RMSF | 11.44 Å | 3.12 Å | 1.24 Å |
注:TM 分数是衡量两条蛋白质结构在全局拓扑上的相似度的归一化指标,通过在各种可能的对齐方式中最大化长度归一的结构重合度计算得到,取值 0–1,越接近 1 表示整体折叠越相似。
一般在结构生物学和结构比对的经验里,TM-score ≈ 0.5 是一个比较公认的折叠类型分界线。所以本项目选取介于最发散构象的最低TM分数,在低于0.5和高于0.5的两个蛋白分别实验,并取具有明确路径的蛋白来做结果验证。从ATLAS数据库中选取每个蛋白的3次重复MD轨迹中的R1轨迹(10,000 frames)作为实验示例。
(二)无向图构建结果分析
1. 聚类数量选择
1.1 选择方法
展示三个蛋白
对各个节点数下的图的统计值valid_paths_count,n_critical_nodes综合并评分。
其中valid_paths_count表示图中从起点到终点的可行路径中总代价小于1.5倍最优路径总代价的个数。
n_critical_nodes表示图中存在的关键节点个数,即可行路径中出现次数前 20% 的节点数。这些节点是可能存在的关键蛋白质构象。
Total Score计算方式:
步骤一——归一化:
步骤二——加权平均:
当前实现中:
1.2 选择结果
- 2pbi
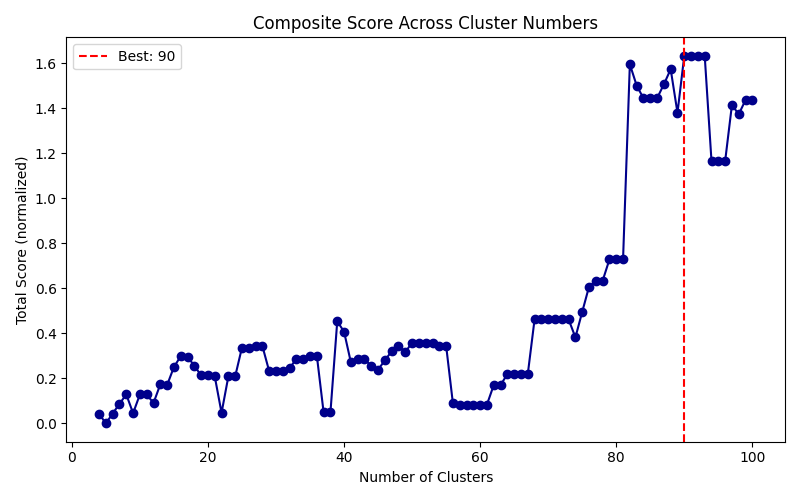
2pbi蛋白4-100节点的图分析结果
- 1k5n
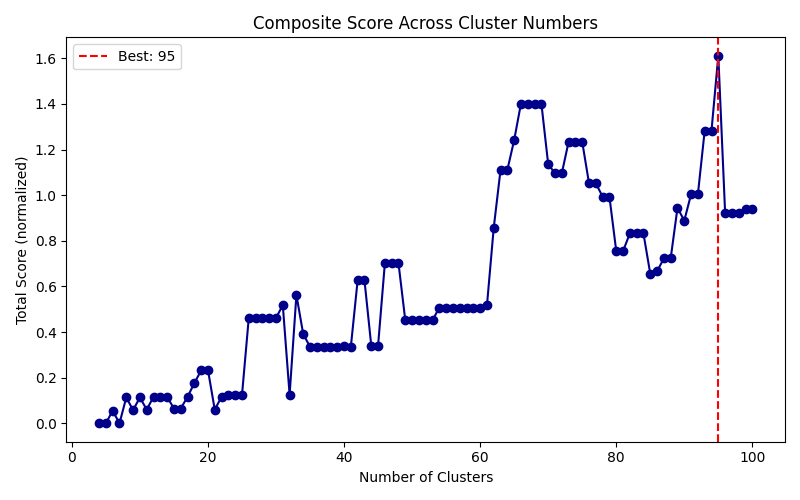
1k5n蛋白4-100节点的分析图
- 2hba
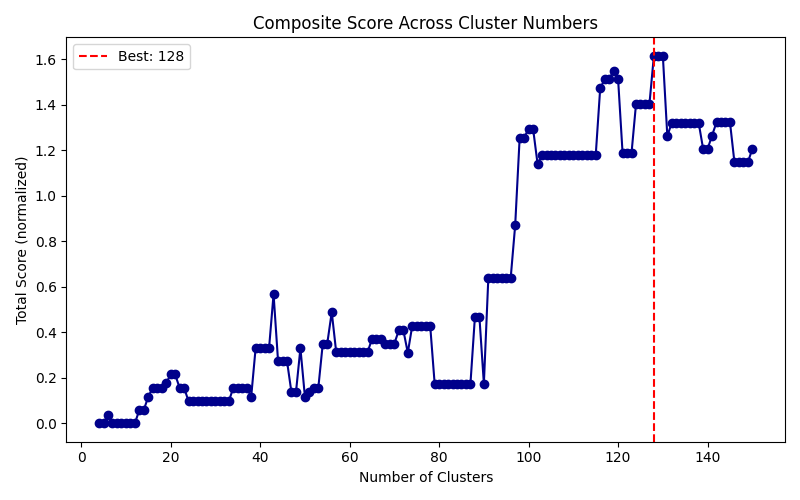
2hba蛋白4-150节点图结构分析图
1.3 选择结论
讨论:根据以上结果可见,在聚类数更多可能在该得分结果下相应的有更好的结果,但相应的计算代价会更高,并且可能不会在后续的关键构象采集中取得更好的效果,所以后续还需综合考虑计算时间和采样效果。考虑到蛋白质在转变过程中的中间稳定状态通常很少,数量仅在个位数,所以更多的中间构象与路径数只会增加计算负担。
2. 确定原始采样方法(FPS/等距)及采样个数
2.1 评测方法
在聚类前序步骤——构象点采样中,FPS采样方法对后续的聚类效果影响很大(在聚类数较少时,路径起点和终点选择为同一个点)。所以对FPS采样、等距采样、全构象集直接聚类进行对比。不进行构象预采样直接聚类的时间复杂度非常高,例如在HC-UPGMA聚类时,计算初始距离矩阵耗时10954.76s,时间代价过大,所以只进行等距采样方法和FPS方法比较。
对于图
1. 有效路径得分:
上式中:
“在所有候选方案上计算”指的是把要比较的所有方案(所有
2. 关键节点得分:(带通评分)
其中:
式中默认
默认权重均为1.0。
根据Score来选择采样个数和聚类节点数,其规则为:
① 固定
② 全局最优找
2.2 评测结果
- 2pbi
待补充
- 1k5n
| n_s | k | valid_paths_count | S_paths | n_critical_nodes | S_crit | score |
|---|---|---|---|---|---|---|
| 200 | 147 | 1.0 | 0.210780 | 6.0 | 0.827586 | 1.038366 |
| 300 | 122 | 32.0 | 1.000000 | 13.0 | 1.000000 | 2.000000 |
| 400 | 88 | 22.0 | 0.953477 | 14.0 | 1.000000 | 1.953477 |
| 500 | 148 | 31.0 | 1.000000 | 7.0 | 0.965517 | 1.965517 |
| 600 | 65 | 12.0 | 0.779979 | 8.0 | 1.000000 | 1.779979 |
| n_s | k | valid_paths_count | n_critical_nodes | S_paths | S_crit | score |
|---|---|---|---|---|---|---|
| 200 | 147 | 503 | 9 | 0.757819 | 1 | 1.757819 |
| 300 | 137 | 566 | 9 | 0.773962 | 1 | 1.773962 |
| 400 | 125 | 3072 | 18 | 1 | 1 | 2 |
| 500 | 118 | 4460 | 20 | 1 | 1 | 2 |
| 600 | 123 | 2813 | 24 | 0.99352 | 1 | 1.99352 |
| n_s | k | valid_paths_count | n_critical_nodes | S_paths | S_crit | score |
|---|---|---|---|---|---|---|
| 200 | 91 | 288 | 28 | 0.802255 | 1 | 1.802255 |
| 300 | 85 | 1008 | 16 | 1 | 1 | 2 |
| 400 | 116 | 850 | 13 | 0.976471 | 1 | 1.976471 |
| 500 | 142 | 1341 | 18 | 1 | 1 | 2 |
| 600 | 85 | 938 | 12 | 0.992345 | 1 | 1.992345 |
| n_s | k | valid_paths_count | n_critical_nodes | S_paths | S_crit | total_score |
|---|---|---|---|---|---|---|
| 200 | 138 | 13518 | 25 | 1 | 1 | 2 |
| 300 | 126 | 10704 | 31 | 1 | 1 | 2 |
| 400 | 137 | 9411 | 16 | 1 | 1 | 2 |
| 500 | 142 | 403 | 7 | 0.628763 | 1 | 1.628763 |
| 600 | 109 | 256 | 18 | 0.575184 | 1 | 1.575184 |
- 2hba
| n_s | k | valid_paths_count | n_critical_nodes | S_paths | S_crit | total_score |
|---|---|---|---|---|---|---|
| 200 | 132 | 61 | 18 | 0.977043 | 1 | 1.977043 |
| 300 | 136 | 26 | 19 | 0.780245 | 1 | 1.780245 |
| 400 | 87 | 72 | 12 | 1 | 1 | 2 |
| 500 | 98 | 128 | 17 | 1 | 1 | 2 |
| 600 | 117 | 114 | 19 | 1 | 1 | 2 |
| n_s | k | valid_paths_count | n_critical_nodes | S_paths | S_crit | total_score |
|---|---|---|---|---|---|---|
| 200 | 126 | 16 | 19 | 1 | 1 | 2 |
| 300 | 39 | 4 | 3 | 0.647685 | 1 | 1.647685 |
| 400 | 140 | 9 | 19 | 0.926628 | 1 | 1.926628 |
| 500 | 68 | 5 | 4 | 0.721057 | 1 | 1.721057 |
| 600 | 7 | 1 | 1 | 0.278943 | 1 | 1.278943 |
| n_s | k | valid_paths_count | n_critical_nodes | S_paths | S_crit | total_score |
|---|---|---|---|---|---|---|
| 200 | 110 | 30 | 9 | 1 | 1 | 2 |
| 300 | 127 | 10 | 18 | 0.664632 | 1 | 1.664632 |
| 400 | 138 | 10 | 18 | 0.664632 | 1 | 1.664632 |
| 500 | 141 | 30 | 14 | 1 | 1 | 2 |
| 600 | 79 | 5 | 6 | 0.428317 | 1 | 1.428317 |
2.3 评测结论
- 采样方式:在样本量较低(n_s≈200)时,等距采样显著优于FPS。FPS 在 1k5n 的 200 构象下更易偏向高 RMSD 区域,导致层次聚类(尤其 UPGMA)把高/低 RMSD 混成一簇,起终点被选为同一点,路径不可用或“假最短”概率上升。
- 聚类算法稳定性:
- KMeans 在两套蛋白上都表现最稳,在 n_s=400–600 时多次达到 score≈2(满分)。
- UPGMA 对采样分布更敏感:1k5n 在等距采样+中高 n_s 时能达优;2hba 的表现起伏更大(n_s=200 可达 2,但 n_s=600 明显下滑)。
- UPGMC 在 1k5n 的低 n_s(200–400)下能拿到高分,但在更高 n_s(500–600)开始退化;在 2hba 上也呈现“有时满分、有时回落”的不稳定特征。
- 超参数比例规律:当n_s在400–600区间、k约为n_s的20%–30% 时,最容易得到 S_paths≈1、S_crit≈1(总分≈2) 的优解。
- 关键节点数(n_critical_nodes):优解通常对应 1k5n:~18–24个;2hba:~12–20个。这与“路径骨架”足够稀疏以保证可达性、又不过密以避免自相矛盾”相吻合。
各蛋白的“推荐配置”
(优先顺序:等距采样 > KMeans;若想横向对比再试 UPGMA/UPGMC)
1) 1k5n
- 首选:等距采样 + KMeans
- n_s=400, k≈125(≈31%)→ 记分≈2.0
- n_s=500, k≈118(≈24%)→ 记分≈2.0
- n_s=600, k≈123(≈20%)→ 记分≈1.99
- 备选:等距采样 + UPGMA
- n_s=300, k≈85 或 n_s=500, k≈142 → 记分≈2.0
- 不建议:FPS@200 尤其配层次聚类(UPGMA)——容易起终点重合、失真。
2) 2hba
- 首选:等距采样 + KMeans
- n_s=400, k≈87(≈22%)→ 记分≈2.0
- n_s=500, k≈98(≈20%)→ 记分≈2.0
- n_s=600, k≈117(≈20%)→ 记分≈2.0
- 备选:
- 等距采样 + UPGMA(n_s=200, k≈126) → 记分≈2.0(但随 n_s 升高易下滑)
- 等距采样 + UPGMC(n_s=200或 500) → 记分≈2.0(稳定性不如 KMeans)
3. 聚类算法选择
3.1 选择依据
选取10k帧的构象作为总构象集,应用等距采样方法从中采样出200-600个距离分布最远的构象,再分别应用KMeans,HC-UPGMA,HC-UPGMC三种聚类方法聚类出4-100个代表构象。
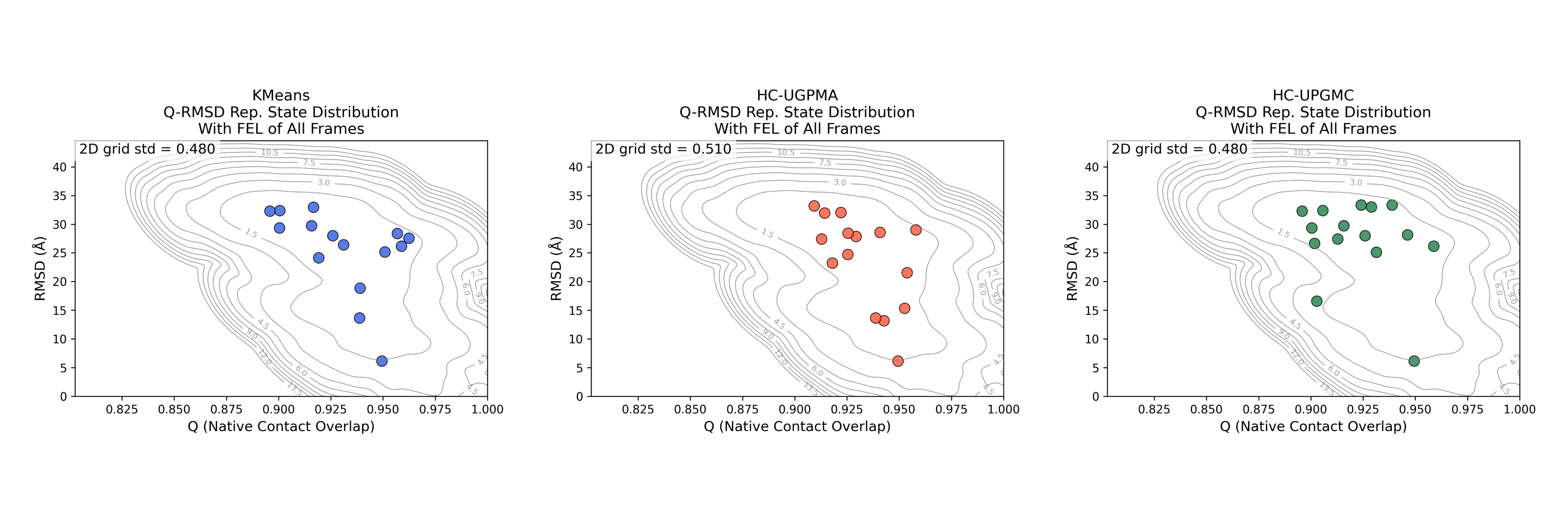
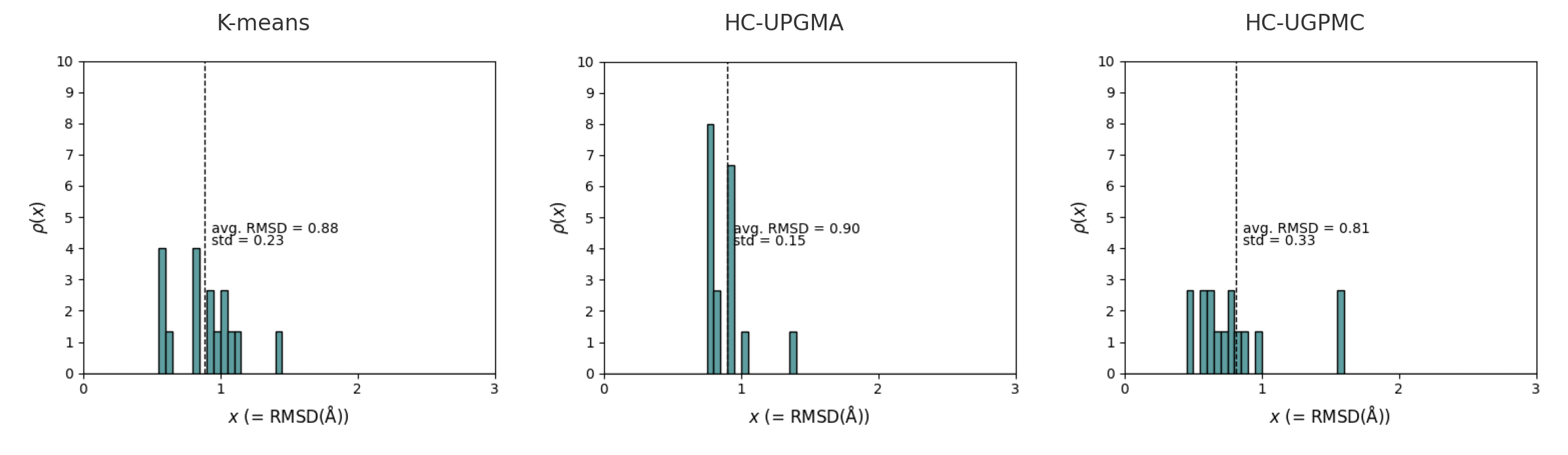
图中上半部分为三种聚类方法下15个代表构象在Q-RMSD能势图中的分布情况。并计算2D网格分布标准差,具体做法为将全构象能到达的Q-RMSD方格作为可及区域,统计代表构象落在各个格子中的个数,得到计数矩阵,计算矩阵中的标准差。这个标准差越低则表示代表构象的分布越均匀,聚类效果越好。
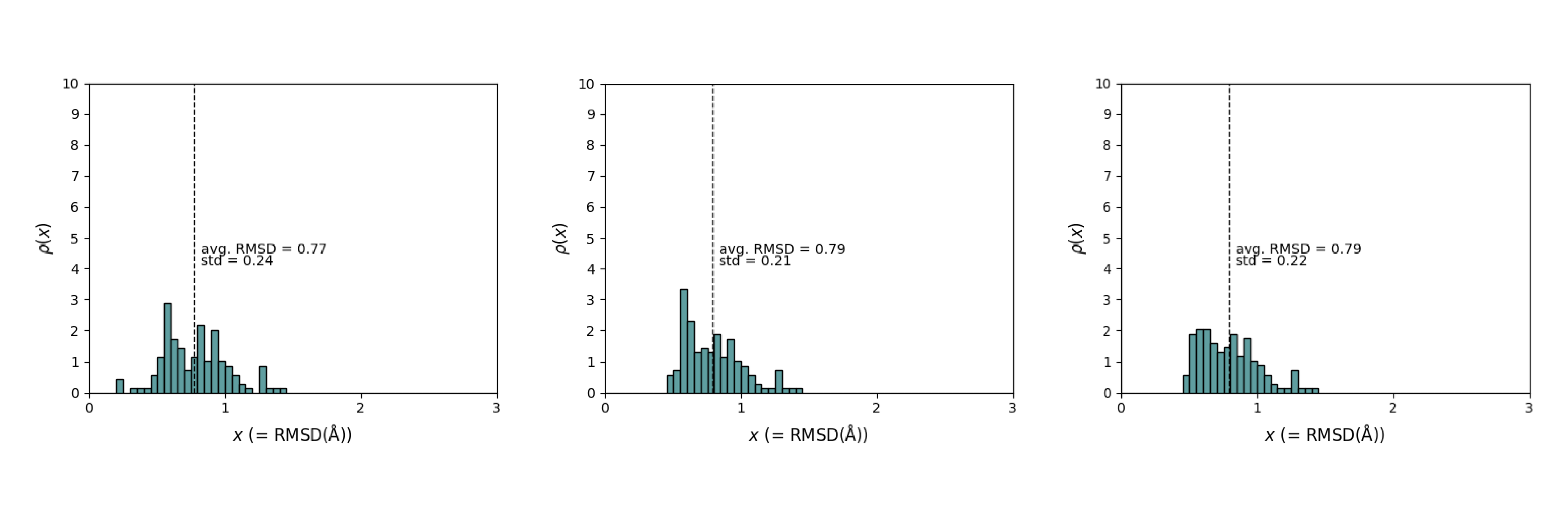
图中下半部分为三种聚类方法下代表构象的最近邻RMSD的分布情况,纵坐标为概率密度。这个分布尽可能的窄的时候表面效果越好。体现在标准差上是最近邻RMSD的标准差越低则分布越均匀,聚类效果越好。
3.3 选择结果
- 2pbi结果
| 方法 | n_s | n_clusters | Nearest Neighbor RMSD std | 2D_grid_std | Score |
|---|---|---|---|---|---|
| KMeans | 200 | 15 | 0.23 | 0.480 | 0.798 |
| HC-UPGMA | 200 | 15 | 0.15 | 0.510 | -0.232 |
| HC-UGPMC | 200 | 15 | 0.33 | 0.480 | -0.566 |
结论:
综合最近邻距离标准差和2D网格分布标准差分析,对于2pbi-A蛋白在15节点时KMeans的聚类效果最好,即代表构象之间分布最广且相隔距离平均。
- 1k5n结果
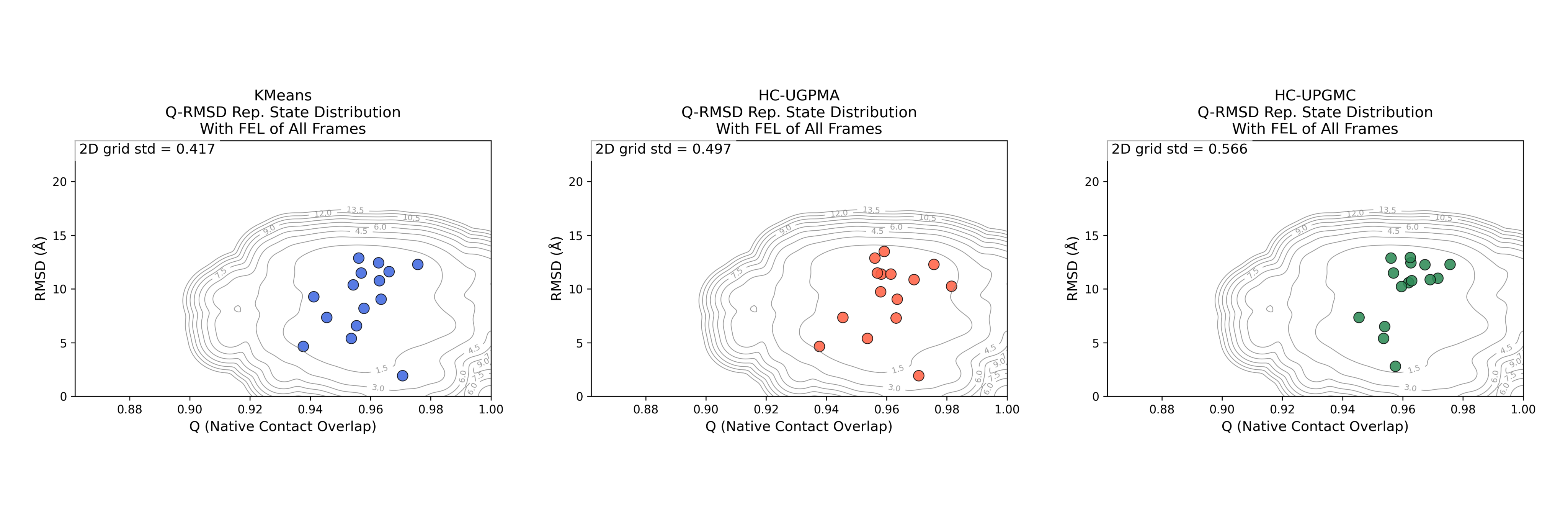
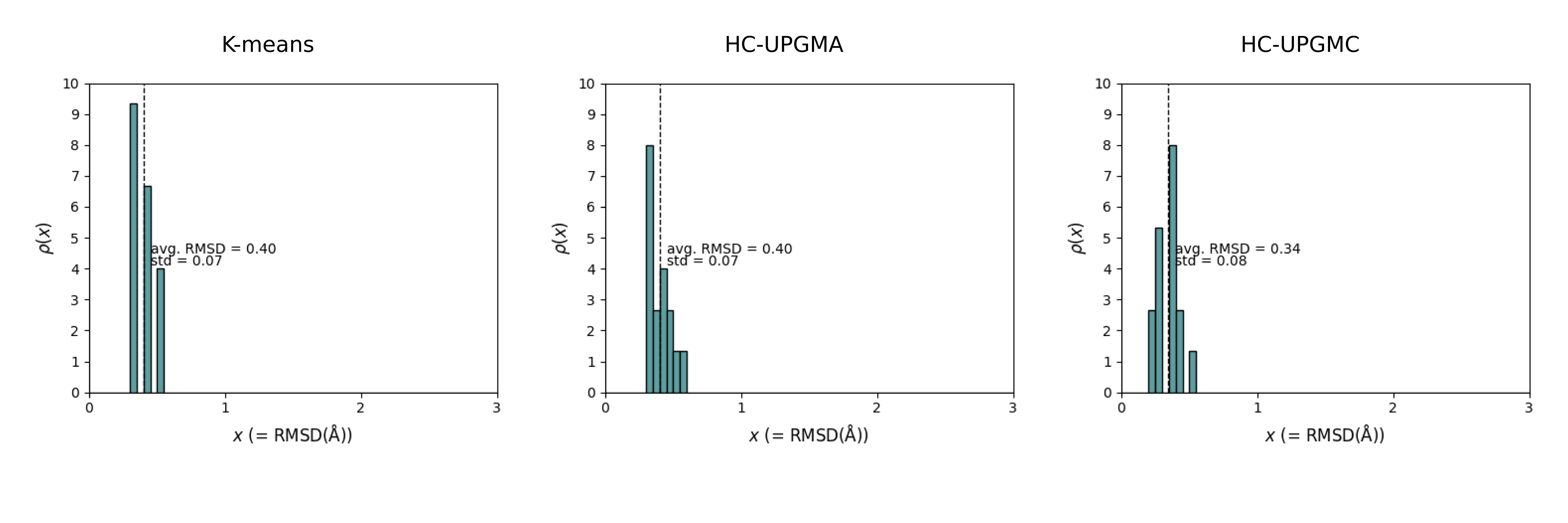
| 方法 | n_s | n_clusters | Nearest Neighbor RMSD std | 2D_grid_std | Score |
|---|---|---|---|---|---|
| KMeans | 200 | 15 | 0.07 | 0.417 | 1.983 |
| HC-UPGMA | 200 | 15 | 0.07 | 0.497 | 0.585 |
| HC-UPGMC | 200 | 15 | 0.08 | 0.556 | -2.564 |
结论:对于1k5n-A蛋白,在15个节点情况下,KMeans的聚类效果最好。(可能受限于节点个数,需进一步测试)
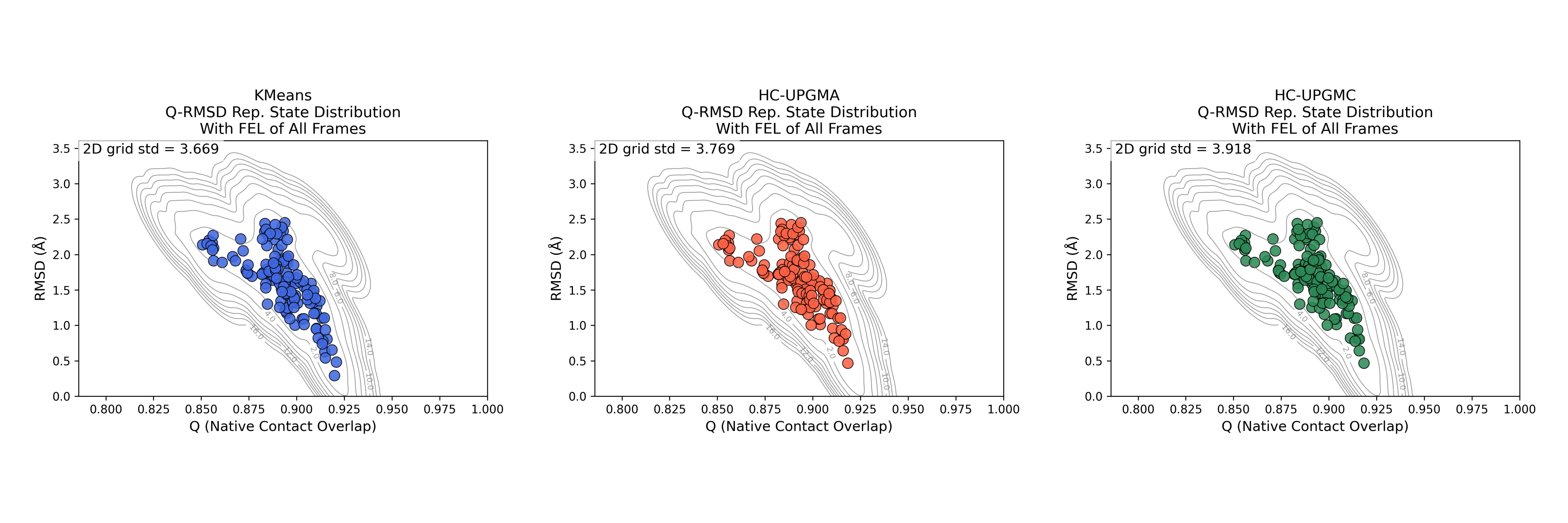
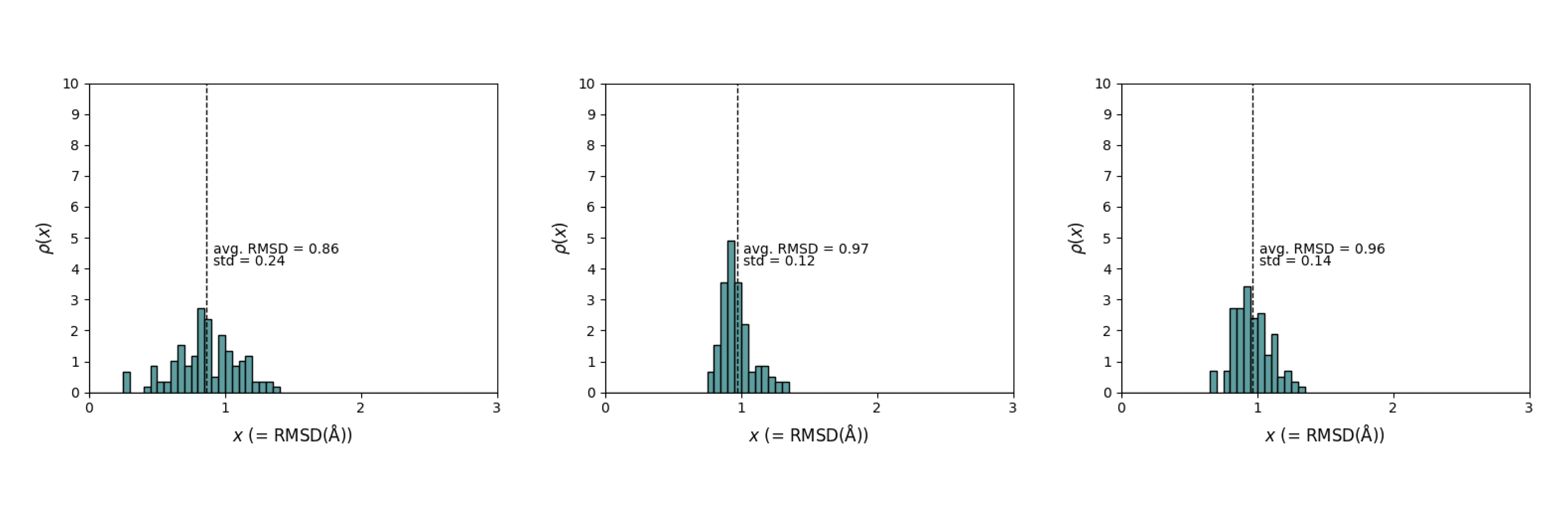
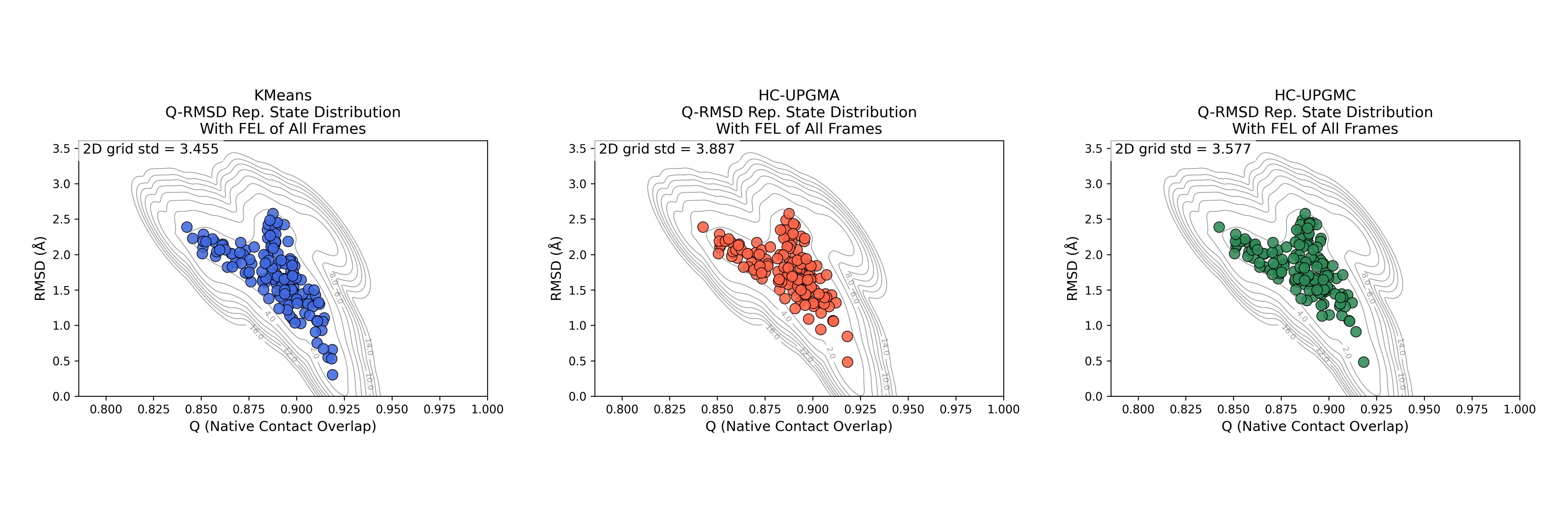
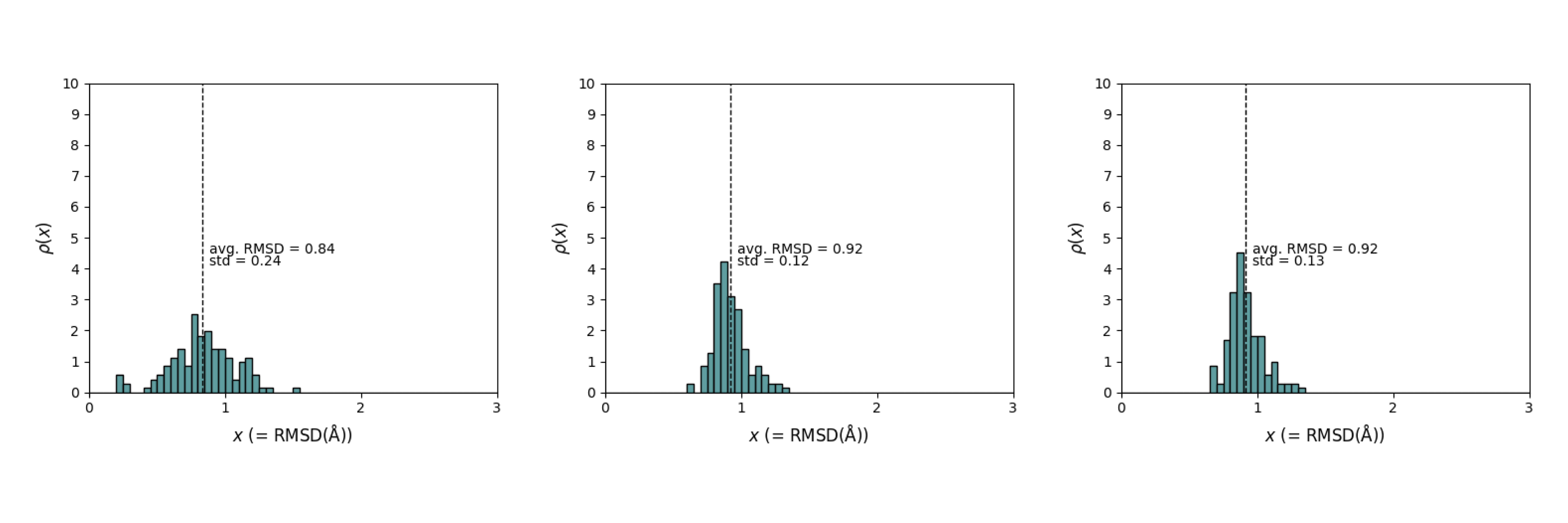
| Method | n_s | n_clusters | Nearest Neighbor RMSD std | 2D_grid_std | Score |
|---|---|---|---|---|---|
| KMeans | 200 | 138 | 0.238569 | 3.66913 | -0.136799 |
| UPGMA | 200 | 138 | 0.214284 | 3.769299 | 0.472309 |
| UPGMC | 200 | 138 | 0.216057 | 3.918234 | -0.335510 |
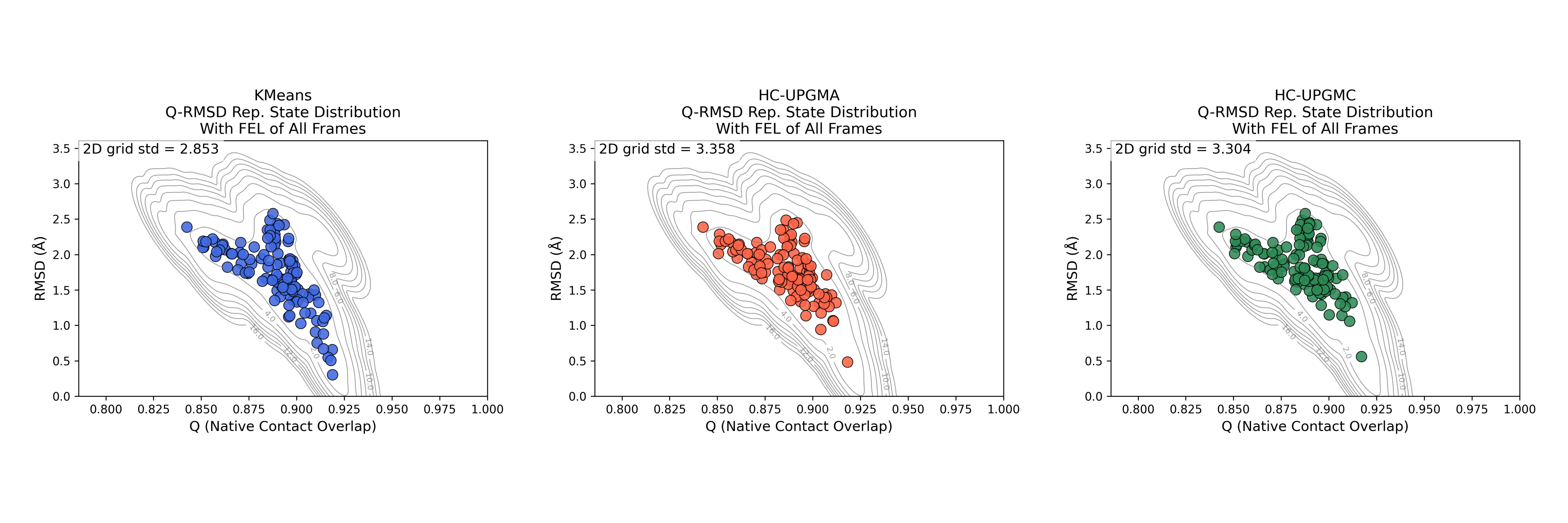
| KMeans | 500 | 118 | 0.236632 | 2.85252 | 0.003414 |
| UPGMA | 500 | 118 | 0.117603 | 3.357696 | 0.023185 |
| UPGMC | 500 | 118 | 0.135357 | 3.303558 | −0.026599 |
| KMeans | 500 | 142 | 0.235319 | 3.454544 | −0.198715 |
| UPGMA | 500 | 142 | 0.124873 | 3.887202 | -0.323416 |
| UPGMC | 500 | 142 | 0.125466 | 3.577236 | 0.522131 |
注:Score计算方法:首先对两种std值分别进行Z-Score标准化。
其中
结论:
1k5n蛋白三种聚类方法的结果来看,整体上HC-UPGMA方法要优于KMeans与HC-UPGMC方法。在平均最近邻RMSD标准差的指标上,UPGMA的效果最优,在分布上表现为数据分布最窄最集中。
- 2hba结果:






| Method | n_s | n_clusters | Nearest Neighbor RMSD std | 2D_grid_std | Score |
|---|---|---|---|---|---|
| KMeans | 600 | 117 | 0.391484 | 3.413684 | -0.136394 |
| UPGMA | 600 | 117 | 0.173797 | 4.004524 | 0.520492 |
| UPGMC | 600 | 117 | 0.168829 | 4.532864 | -0.384098 |
| KMeans | 200 | 126 | 0.41082619 | 4.40554 | -0.266441 |
| UPGMA | 200 | 126 | 0.35071598 | 4.453573 | 1.06431 |
| UPGMC | 200 | 126 | 0.366571103 | 4.562131 | -0.797866 |
| KMeans | 500 | 141 | 0.372809 | 4.594168 | -0.026673 |
| UPGMA | 500 | 141 | 0.193696 | 5.275395 | -0.112528 |
| UPGMC | 500 | 141 | 0.216549 | 5.10319 | 0.1392 |
结论:2hba蛋白三种聚类方法的结果来看,部分的数据上HC-UPGMA方法要优于KMeans与HC-UPGMC方法。在平均最近邻RMSD标准差的指标上,UPGMA的效果最优,在分布上表现为数据分布最窄最集中。所以在选择聚类方法时,可根据具体蛋白测试后,具体选择相应方法。
4. 与起点和终点连接边数分析
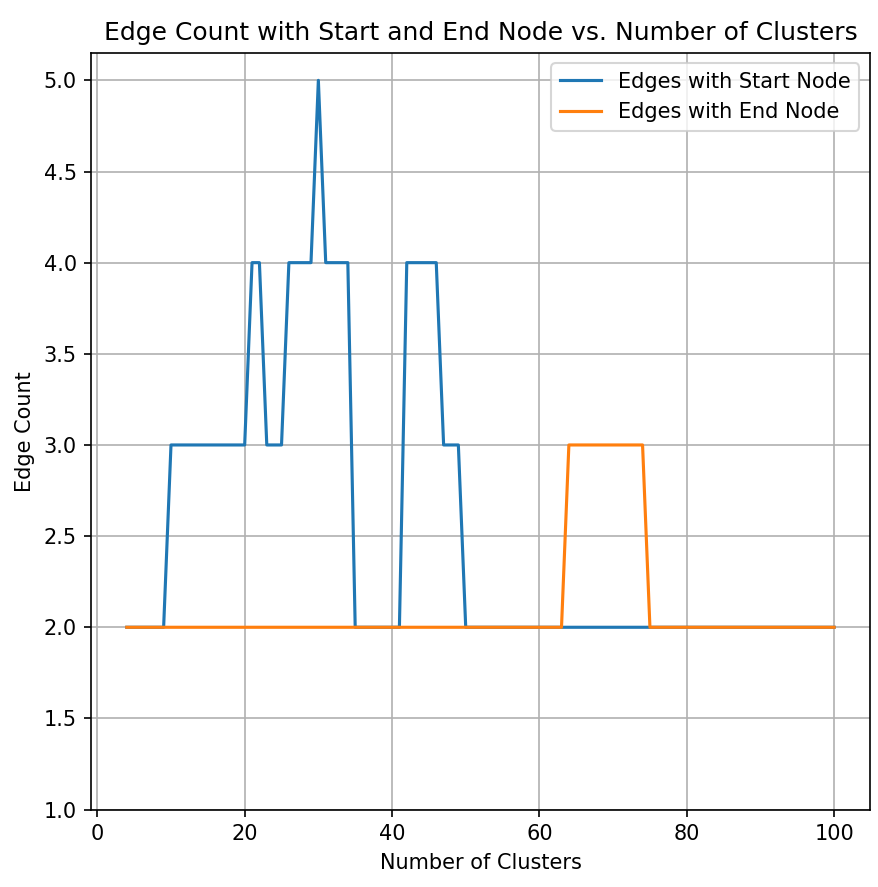
结论:在4-100个代表构象的图构建结果中,与起点和终点的连接边数很少(均不大于5)。后续可以尝试在建模的时候优化约束数量。
(三)QUBO模型的惩罚系数与SA最优参数组合的搜索结果
1、搜索方法
在此处对不同蛋白的采样参数设置进行网格搜索,寻找建模的最优惩罚系数以及模拟退火的最优参数组合。对于参数组合的得分评价采用以下定义:
其中
2、搜索结果
| 蛋白 | 节点数 | Ls | Lt | Lr | i_t | alpha | per_t |
|---|---|---|---|---|---|---|---|
| 2pbi | 15 | 200 | 300 | 700 | 1000 | 0.65 | 5000 |
| 2pbi | 20 | 100 | 200 | 900 | 1000 | 0.75 | 5000 |
| 2pbi | 25 | 400 | 400 | 1100 | 1000 | 0.65 | 25000 |
| 2pbi | 30 | 900 | 400 | 1300 | 1000 | 0.75 | 10000 |
| 2pbi | 35 | 400 | 100 | 700 | 1000 | 0.6 | 25000 |
| 1k5n | 15 | 700 | 800 | 1300 | 1000 | 0.8 | 5000 |
| 1k5n | 20 | 600 | 500 | 1300 | 1000 | 0.6 | 5000 |
| 1k5n | 25 | 1000 | 600 | 1700 | 1000 | 0.75 | 15000 |
| 1k5n | 30 | 1000 | 1000 | 3000 | 1000000 | 0.95 | 30000 |
| 1k5n | 35 | 1000 | 1000 | 1700 | 10000000 | 0.95 | 30000 |
| 2hba | 15 | 1000 | 1000 | 1700 | 1000 | 0.7 | 5000 |
| 2hba | 20 | 1000 | 1000 | 1900 | 1000 | 0.6 | 5000 |
| 2hba | 25 | 1000 | 1000 | 2000 | 1000 | 0.75 | 5000 |
注:其中Ls,Lt,Lr分别为起点、终点、中间节点约束强度,i_t为初始温度,alpha为衰减系数,per_t为每个温度下的迭代次数。
示例:
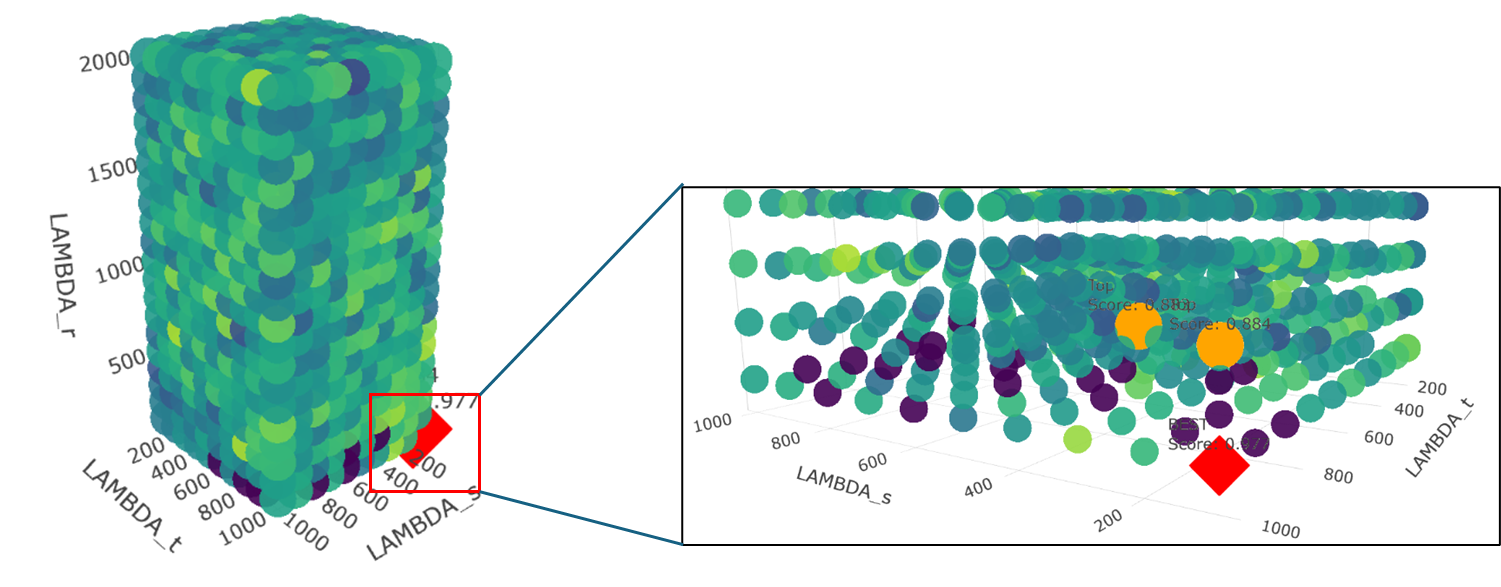
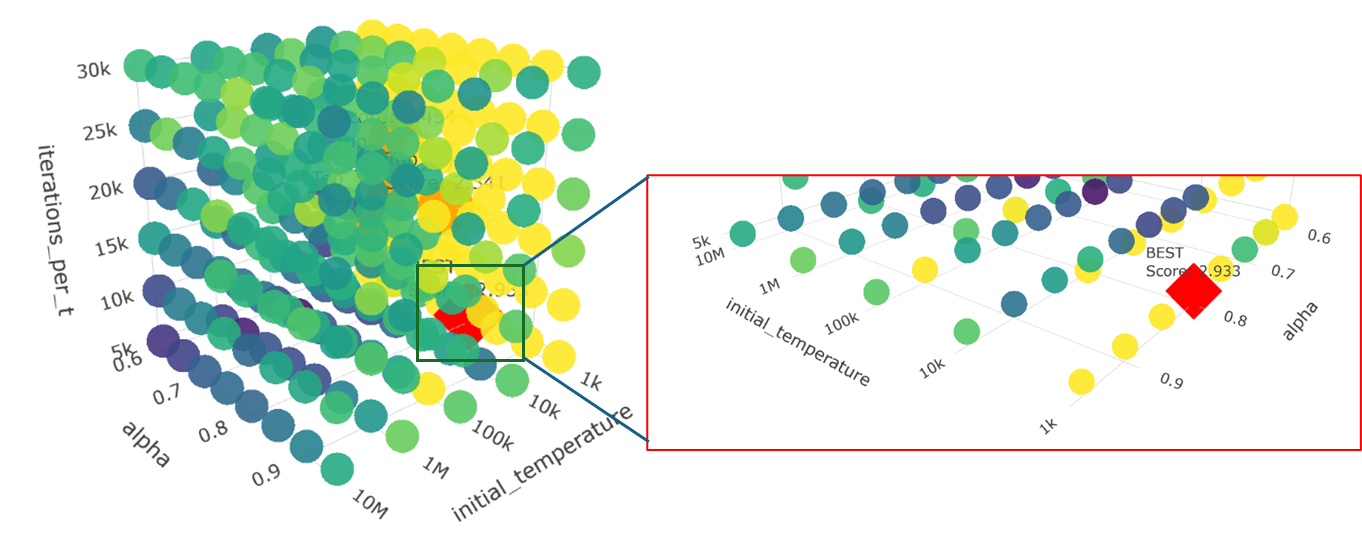
| 蛋白 | 节点数 | Ls | Lt | Lr | i_t | alpha | per_t | 采样路径数 |
|---|---|---|---|---|---|---|---|---|
| 2pbi | 15 | 800 | 1000 | 2000 | 1000 | 0.6 | 20000 | 4 |
| 2pbi | 20 | 100 | 200 | 900 | 10000000 | 0.95 | 20000 | 16 |
| 2pbi | 25 | 400 | 400 | 1100 | 10000 | 0.95 | 25000 | 8 |
| 2pbi | 30 | 900 | 400 | 1300 | 10000 | 0.95 | 30000 | 27 |
| 2pbi | 35 | 400 | 100 | 700 | 100000 | 0.95 | 30000 | 25 |
| 1k5n | 15 | 700 | 800 | 1300 | 10000000 | 0.95 | 30000 | 59 |
| 1k5n | 20 | 500 | 800 | 900 | 1000 | 0.95 | 30000 | 24 |
| 1k5n | 25 | 1000 | 900 | 1800 | 100000 | 0.95 | 30000 | 27 |
| 1k5n | 30 | 600 | 800 | 1400 | 1000 | 0.95 | 25000 | 17 |
| 1k5n | 35 | 200 | 800 | 900 | 10000000 | 0.95 | 30000 | 29 |
| 2hba | 15 | 100 | 1000 | 1900 | 1000 | 0.95 | 15000 | 4 |
| 2hba | 20 | 1000 | 1000 | 2000 | 1000 | 0.6 | 5000 | 2 |
| 2hba | 25 | 1000 | 1000 | 2000 | 1000 | 0.75 | 5000 | 2 |
注:基于相干伊辛机的量子技术在计算方面的优势远超经典计算机,所以在这里统计了最多采样路径数的参数组合。
结论:
当目标是在较短探索时间上获得较多路径,则采用1000的初始温度,并采用较低或中等水平(5000-15000)的迭代次数(i_t),退火衰减系数(alpha)多集中在0.6–0.8之间,
当目标是最大化路径多样性,则应采用高初始温度、高alpha、高迭代次数的组合,并且在惩罚系数的选择上,Lr通常在Lt的1.5~2倍,Ls则在Lt的0.9-1.5倍选择。
从整体优化结果可以得知,即使惩罚系数理论上应该是远大于
在2hba的20和25节点规模的参数网格搜索中,发现参数都达到网格搜索空间的最大边界。表明可能约束强度不足,应使用更大的搜索空间。
(四)路径采样结果分析
1、分析结果
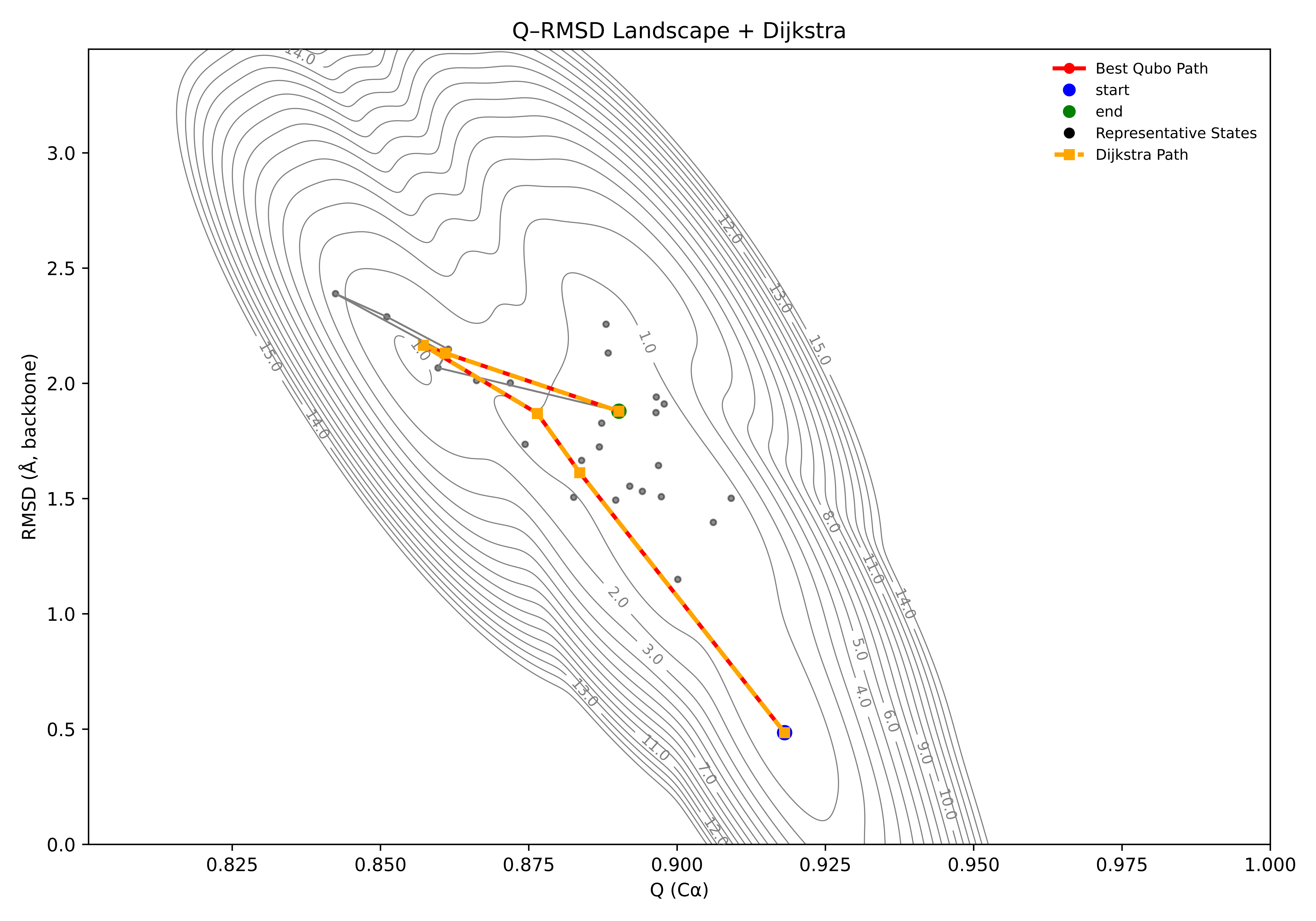
本项目方法对2pbi蛋白在20个节点规模下在上述最优参数下所有可行路径与Dijkstra路径如图显示:
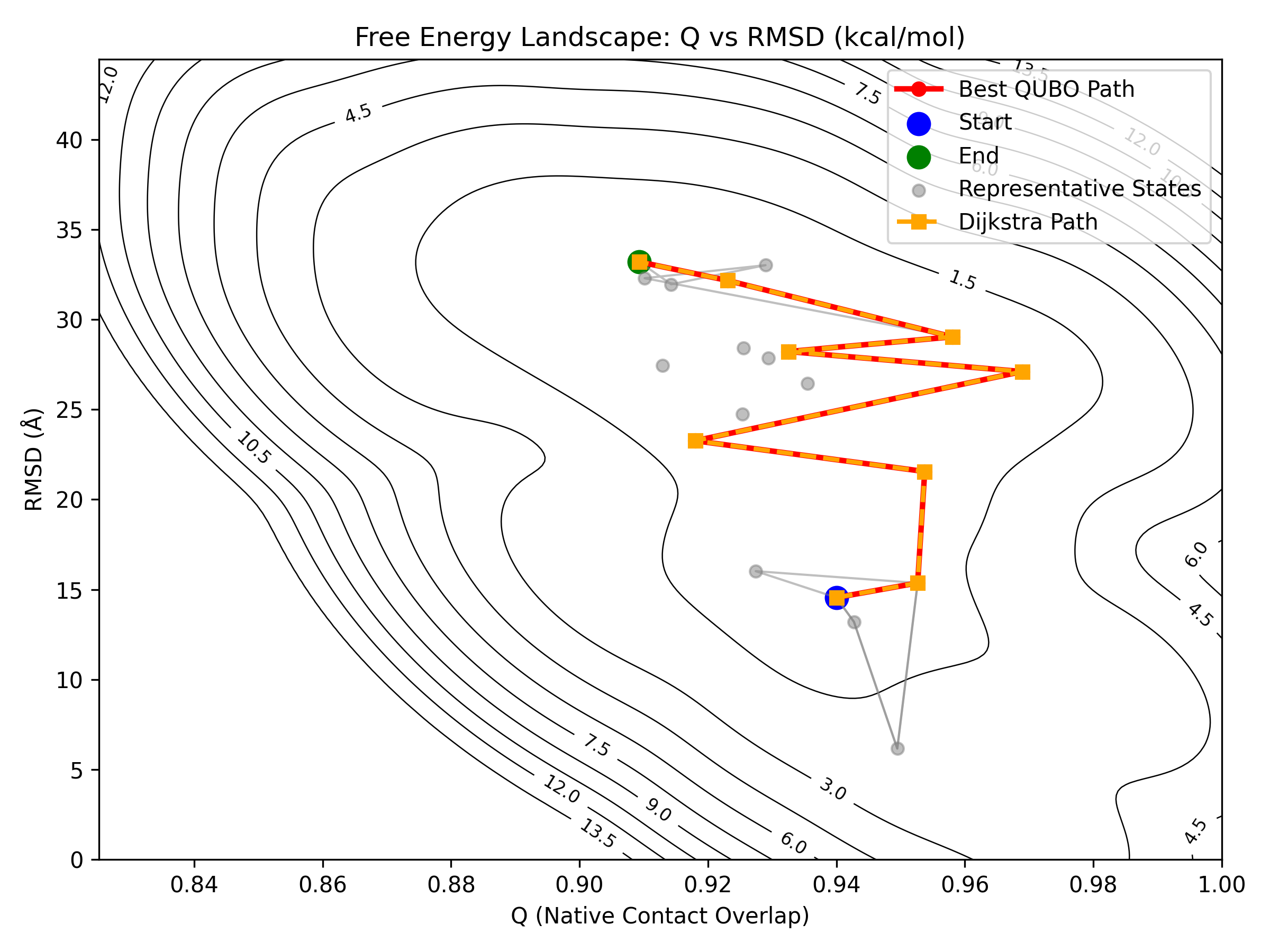
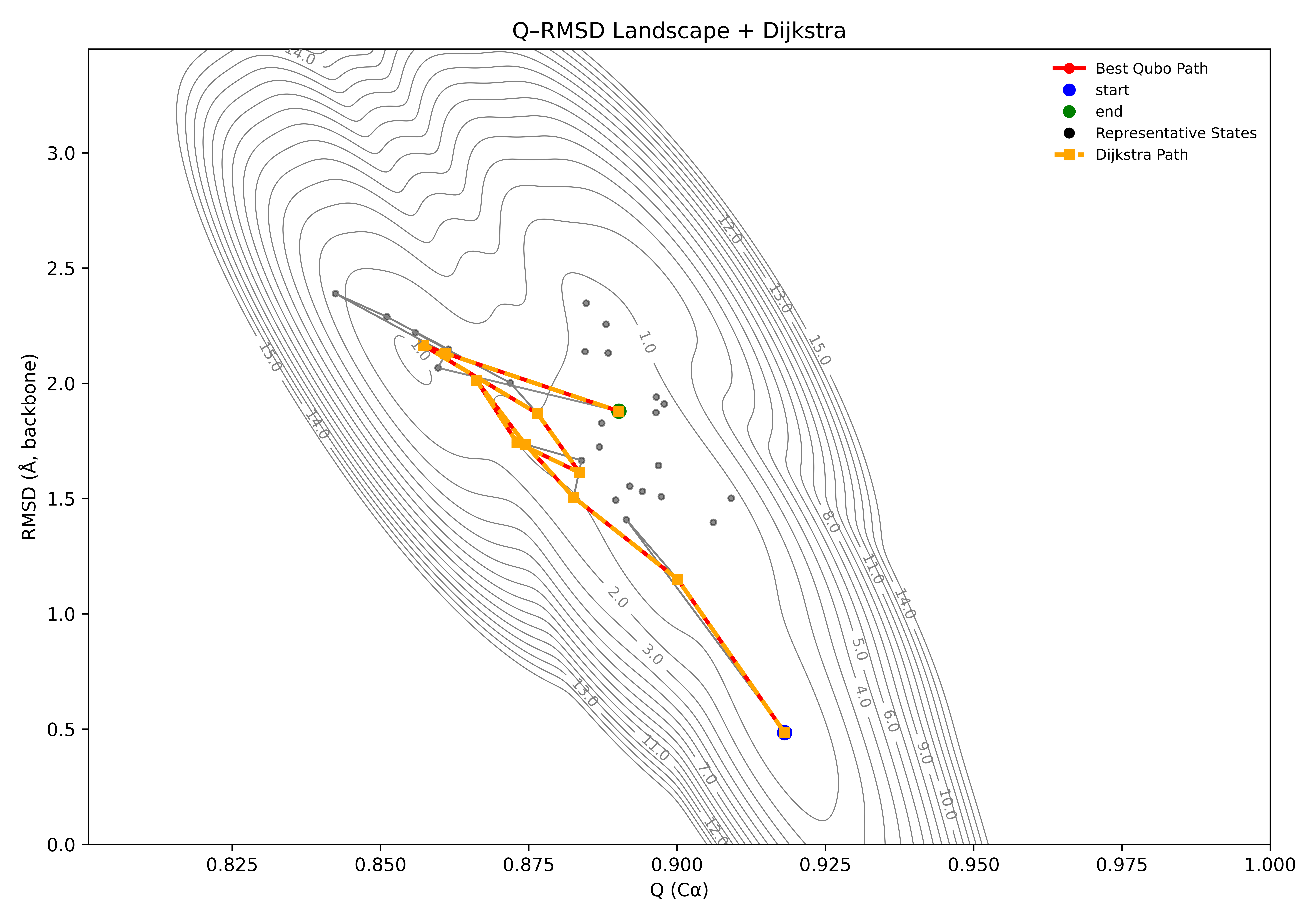
在更高的SA参数下(得到最多可行解数:“11”个)的路径采样结果显示:
(initial temprature = 1e7,alpha = 0.95,iterations_per_t=20K)
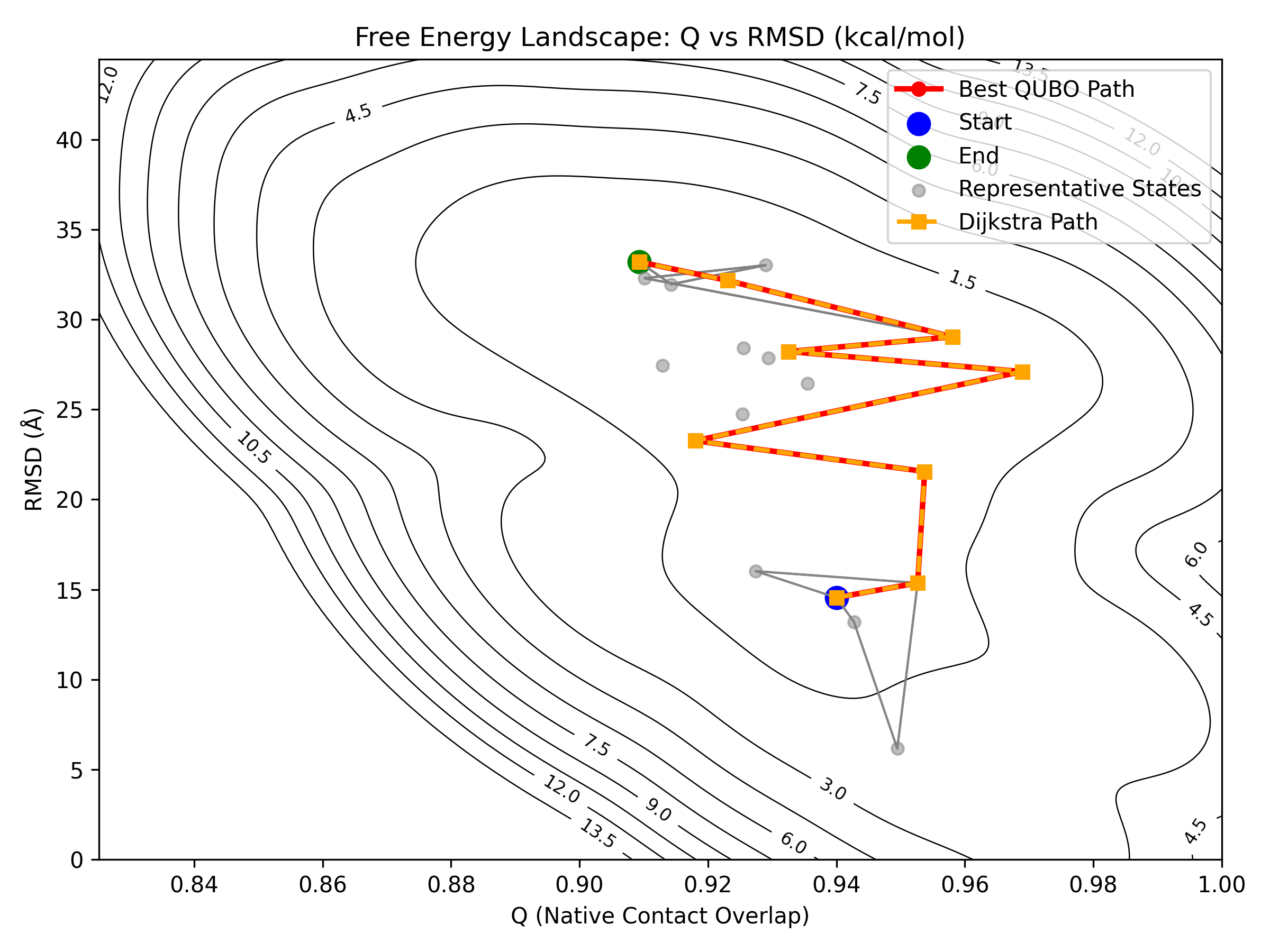
结论:本方法可以很好的采样出最优路径,并且可以很好的采样出较多次优路径,并识别出其中的关键构象。两个SA参数相比之下可以发现,计算深度较浅时也可以采出较全的可行路径。
2、分析结论
最优性:在合适参数下,方法能稳定命中 Dijkstra 全局最优路径。
多样性:通过增大 SA 参数(或等价的探索强度),可显著提升次优路径的采样覆盖度,便于做路径稳健性/冗余性分析。
关键态识别:在多路径框架中,**关键构象(瓶颈/关节点)**能被反复命中,具有良好可复现性。
计算效率:在 2pbi 的对比中,较浅搜索深度已能采到较完整的可行解集,支持分阶段/分层探索策略:先浅后深、先粗后细。
节点规模影响:适度提高节点规模(如 1k5n 的 30→35)有利于细化能势通道与次优通路,但不会改变最优结论;因此可按需求在分辨率与计算成本间权衡。
(五)经典TPS方法实验
1、实验过程
对2pbi蛋白n_clusters = 20(因为可行路径数更多,有6条)
原图中:
【最小总权重路径】
- 路径: [2, 10, 11, 18, 17, 16, 6, 7, 4]
- 总权重: 108.93196759854607
【所有总权重不超过最小总权重两倍的路径】
路径: [2, 1, 0, 10, 11, 18, 17, 16, 6, 9, 8, 5, 4],总权重: 170.1089695166429
路径: [2, 1, 0, 10, 11, 18, 17, 16, 6, 7, 4],总权重: 151.3363589604736
路径: [2, 10, 11, 18, 17, 16, 6, 9, 8, 5, 4],总权重: 127.70457815471532
路径: [2, 10, 11, 18, 17, 16, 6, 7, 4],总权重: 108.93196759854607
路径: [2, 3, 10, 11, 18, 17, 16, 6, 9, 8, 5, 4],总权重: 146.9568004285432
路径: [2, 3, 10, 11, 18, 17, 16, 6, 7, 4],总权重: 128.18418987237396
共计6条路径。
应用TPS(“Transition Path Sampling”):
采用对路径中间节点的邻居随机游走扰动方法,产生新路径。
在重复优化路径1e6次的情况下,采出4条可行路径,重复运行3次:
rep1:
Path 1: [2, 3, 10, 11, 18, 17, 16, 6, 7, 4], Weight: 128.18418987237396
Path 2: [2, 3, 10, 11, 18, 17, 16, 6, 7, 4], Weight: 128.18418987237396
Path 3: [2, 10, 11, 18, 17, 16, 6, 7, 4], Weight: 108.93196759854607
rep2:
Path 1: [2, 10, 11, 18, 17, 16, 6, 7, 4], Weight: 108.93196759854607
Path 2: [2, 3, 10, 11, 18, 17, 16, 6, 9, 8, 5, 4], Weight: 146.9568004285432
Path 3: [2, 3, 10, 11, 18, 17, 16, 6, 9, 8, 5, 4], Weight: 146.9568004285432
Path 4: [2, 10, 11, 18, 17, 16, 6, 9, 8, 5, 4], Weight: 127.70457815471532
rep3:
Path 1: [2, 1, 0, 10, 11, 18, 17, 16, 6, 9, 8, 5, 4], Weight: 170.1089695166429
2、实验结论
该方法依赖初始值,且由于是对中间节点的邻居进行的随机游走扰动,采样路径之间的相关性很强。求解不稳定,无法每次得到最优路径,也无法完整采样出所有可能存在的蛋白质转变路径。
(六)路径采样结果验证
基于2hba蛋白已有先验的转变路径与中间折叠状态Seffernick JT, Ren H, Kim SS, Lindert S. Measuring Intrinsic Disorder and Tracking Conformational Transitions Using Rosetta ResidueDisorder.,所以本项目方法对2hba蛋白在2**5**个节点规模下在上述最优参数下所有可行路径与Dijkstra路径如图显示:
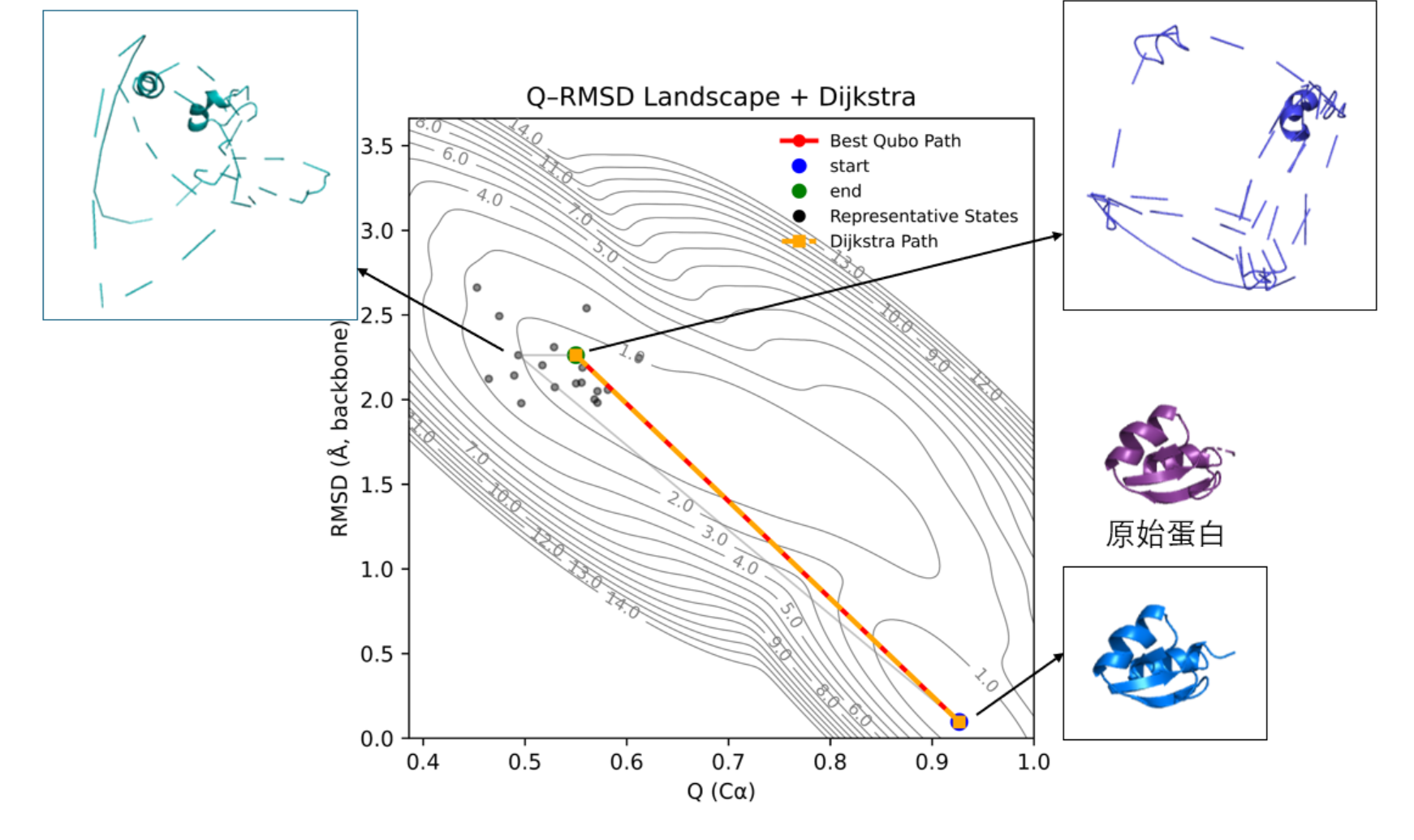
结论:对于2hba蛋白可以采样出转变路径中可能存在的中间结构。可以看出找出的关键中间构象处于螺旋和折叠局部已经部分松动,但整体骨架还维持住的状态。有局部二级结构(短α-helix、小环)还存在,但很多接触丢失;主链整体有明显偏离原始状态的趋势。是处于N 端 α-helix 部分解开 + 局部接触丢失的中间态。
右上角所示的终点构象主体框架已经明显松散,整体 RMSD 比 native 态大很多;只有一段较稳定的 短 α-helix(右上角片段),其余部分基本变成无规卷曲或局部残余 β-turn;很多长程接触完全丢失,只剩下 backbone 支撑的大框架。是处于蛋白质原始状态和去折叠状态转换路径上的高能中间态。
所以本项目可以很好的找出蛋白质转变的路径,当有数量足够多的构象集时,可以更加清晰的找出大分子构象的转变路径以及可能存在的中间状态以便后续的分析。
五、讨论与展望
本研究围绕大分子(蛋白质)构象转移路径的建模与采样问题,实现了基于 QUBO 建模与路径优化算法相结合的路径预测框架。在实验层面,结合模拟退火算法(Simulated Annealing, SA)与图结构分析,有效完成了多条构象跃迁路径的探索,并初步验证了该策略在大分子路径采样中的可行性与扩展性。
(一)模拟退火在路径多样性采样中的表现
通过将路径优化问题转化为 QUBO 表达,本研究利用模拟退火算法对该模型进行求解,显示出良好的采样能力。实验表明:
在中小规模图结构下,SA 能够在合理计算时间内高效生成低作用量(action)的可行路径;
多次独立运行结果具有路径多样性,不同路径在结构空间中覆盖分散,重复关键构象节点有限,具备较强的不相关性;
充分说明,本项目方法在蛋白质构象跃迁路径的多通道采样问题中具备天然优势,尤其适用于路径多样性、关键构象识别等目标。
(二)相干伊辛机(CIM)在路径采样中的潜力
尽管SA在当前问题规模下表现稳定,但其在面对更大规模路径空间或更高分辨率图结构时的效率存在瓶颈。为进一步提升路径采样效率与质量,未来将尝试引入相干伊辛机真机计算(Coherent Ising Machine, CIM),以替代传统算法。
(三)未来展望:从模型到硬件的协同进步
本研究的初步结果为使用量子优化思想研究蛋白质动力学提供了可行路径。未来工作将从以下几个方向深入:
QUBO模型优化:进一步设计更合理的目标函数与约束策略,提高模型对复杂路径拓扑的适应性;
量子硬件接入:探索相干伊辛机等物理平台在真实蛋白体系中的路径采样性能;
路径物理解释与生物关联:基于高权重路径进行关键构象识别、功能位点预测;
跨体系验证:将当前路径采样框架推广到RNA折叠、多结构配体结合等复杂分子体系中。
总体而言,随着量子计算与物理模拟平台的快速发展,基于 QUBO 的路径采样方法有望成为蛋白质动力学研究的有力工具,推动生物分子模拟进入高效率、高精度的新阶段。
六、参考文献
[1]Seffernick J T, Ren H, Kim S S, et al. Measuring intrinsic disorder and tracking conformational transitions using Rosetta ResidueDisorder[J]. The journal of physical chemistry B, 2019, 123(33): 7103-7112.
[2]Ghamari D, Hauke P, Covino R, et al. Sampling rare conformational transitions with a quantum computer[J]. Scientific Reports, 2022, 12(1): 16336.
[3] Seffernick JT, Ren H, Kim SS, Lindert S. Measuring Intrinsic Disorder and Tracking Conformational Transitions Using Rosetta ResidueDisorder. J Phys Chem B. 2019 Aug 22;123(33):7103-7112. doi: 10.1021/acs.jpcb.9b04333. Epub 2019 Aug 14. PMID: 31411026; PMCID: PMC6748046.